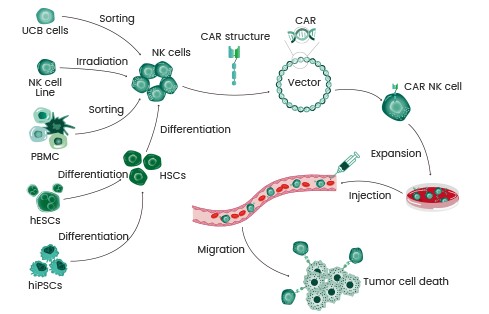
CAR-NK疗法:优势详解与应用前景
CAR-NK细胞疗法作为新型治疗方案,能够克服CAR-T细胞疗法的局限性,如治疗诱导的不良反应。CAR-NK细胞比记忆T细胞和未转导的NK细胞表现出更高的细胞溶解活性;因此,杀伤肿瘤细胞的效果更佳。目前,全球有800多项CAR-T和28项CAR-NK正在开展临床试验。
- 0
- 0
- xiaoqin2023
- 发布于 2023-10-23 16:02
- 阅读 ( 2379 )
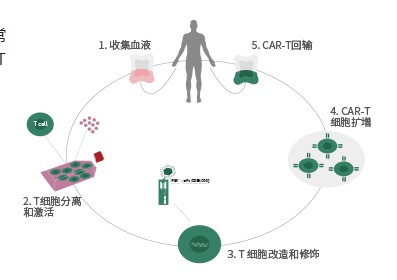
CAR-T疗法:综合概述及临床应用
CAR-T疗法就是嵌合抗原受体T细胞免疫疗法,英文全称Chimeric Antigen Receptor T-Cell Immunotherapy。这是一种治疗肿瘤的新型精准靶向疗法,近几年通过优化改良在临床肿瘤治疗上取得很好的效果,是一种非常有前景的,能够精准、快速、高效,且有可能治愈癌症的新型肿瘤免疫治疗方法。
- 0
- 0
- xiaoqin2023
- 发布于 2023-10-23 15:57
- 阅读 ( 2015 )
Bowtie2使用方法与参数详细介绍
对参考序列构建indexbowtie2-build genome.fasta index尝试使用前10000个reads进行比对bowtie2 -u 10000 -p 8 -x index -1reads1.fq -2 reads2.fq -S out.sam使用8个线程进行比对bowtie2 -p 8...
- 0
- 0
- rzx
- 发布于 2023-10-18 17:58
- 阅读 ( 5072 )
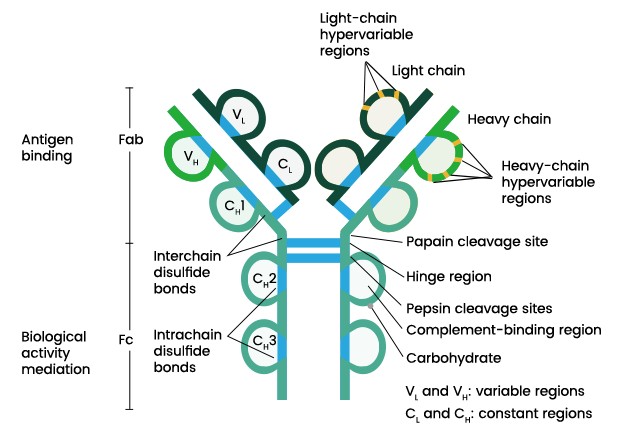
抗体纯化方法概览:四种策略及其优缺点详解
抗体是一类免疫蛋白,可识别并结合特定抗原,在生物研究以及临床应用中,纯化得到高质量的抗体十分重要。
- 0
- 0
- xiaoqin2023
- 发布于 2023-10-17 15:46
- 阅读 ( 3180 )
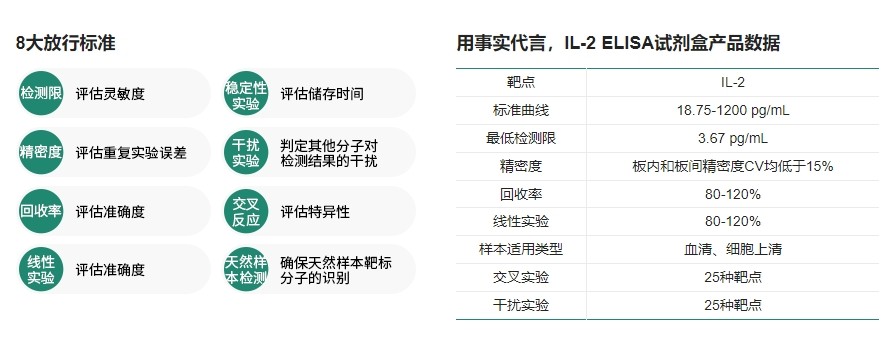
CAR-T质量检测ELISA试剂盒常见问题解析
CAR-T 细胞质量的优劣直接影响临床治疗效果和患者的健康。CAR-T药物的质量在批次和患者间存在差异,因此对CAR-T药物的质量和功能进行表征至关重要。细胞因子释放检测是CAR-T细胞功能检测的重要环节。除此之外,不容忽视的是CAR-T生产工艺过程中引入的杂质检测,如细胞因子残留检测。细胞因子ELISA检测试剂盒可以通过检测代表性细胞因子的表达来评估CAR-T细胞对靶细胞的杀伤活性和特异性。
- 0
- 0
- xiaoqin2023
- 发布于 2023-10-17 15:41
- 阅读 ( 2495 )
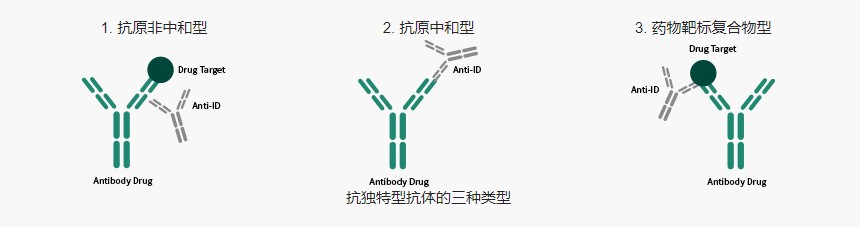
抗独特型抗体制备常见问题解析
抗独特型抗体目前主要用于抗体类药物的血药浓度监测和抗药抗体检测。除此之外,抗独特型抗体也可以作为抗原的“内影像”,用于疫苗开发,在一些自身免疫性疾病、肿瘤、感染性疾病的预防中发挥重要作用。
- 0
- 0
- xiaoqin2023
- 发布于 2023-10-17 15:35
- 阅读 ( 2344 )
BMC genomics|26个玉米基因组中萜烯合酶的多重变异模式
今天给大家分享一篇基于26个玉米泛基因家族文章——“Multiple variation patterns of terpene synthases in 26 maize genomes”
- 0
- 0
- 每天学习一点点
- 发布于 2023-10-16 18:10
- 阅读 ( 3755 )
R包 CandiHap 进行单倍型分析
CandiHap是一款应用于全基因组关联分析(GWAS)后,快速分析基因单倍型来鉴定候选基因的一款友好软件。研究者可通过该软件对单个基因,或者对GWAS显著关联位点LD范围内所有基因进行单倍型分析,发现与性状显著关联的单倍型和候选基因。
- 0
- 0
- Ti Amo
- 发布于 2023-10-16 15:30
- 阅读 ( 6729 )
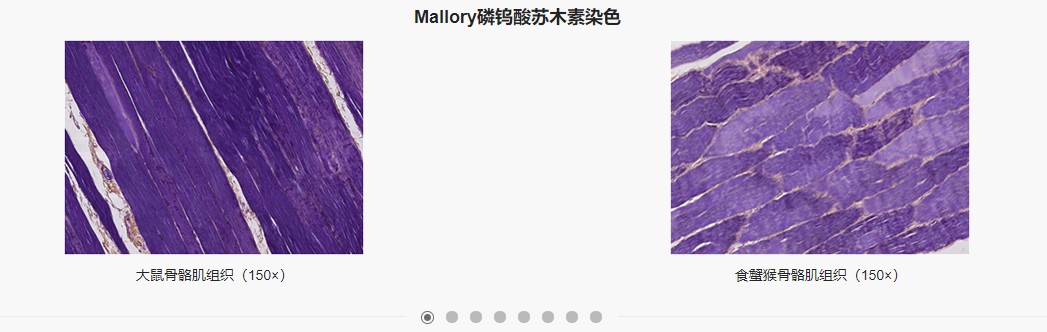
特殊染色技术的原理及其在实践中的应用
HE染色,全称为苏木精-伊红染色法 (hematoxylin-eosin staining),是最基本的病理学染色技术,能够对正常组织和病理组织进行形态结构的观察。特殊染色是常规HE染色的必要补充,能够有针对性地对组织成分进行染色,使组织病理学评价更加完善精确,在临床病理学诊断和病理组织学研究具有重要的应用价值。除常规的HE染色外,义翘神州还提供9种特殊染色服务,包括masson三色染色、 PAS染色、尼氏染色、VG染色、苏丹黑染色、普鲁士蓝染色、油红O染色和β-半乳糖苷酶衰老染色等。
- 0
- 0
- xiaoqin2023
- 发布于 2023-10-13 16:51
- 阅读 ( 4919 )
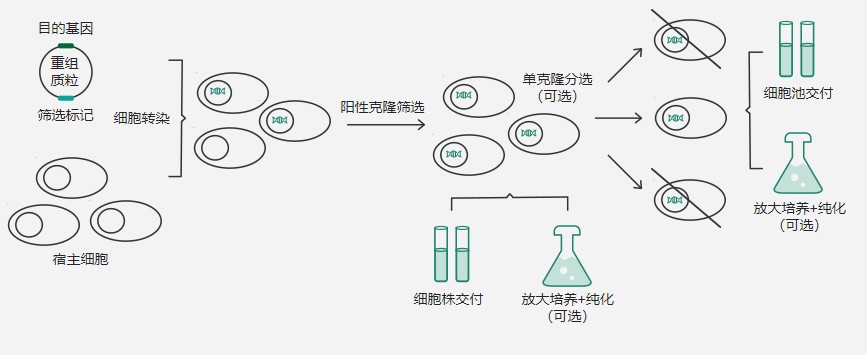
瞬时转染与稳定转染:差异与特点
哺乳动物细胞表达系统具有促使蛋白正确折叠和实现复杂修饰的功能,表达的蛋白更近天然状态,能够运用于制药等活性要求高的领域。利用哺乳动物细胞表达系统生产蛋白通常有两种方式:瞬时转染与稳定转染(构建稳定细胞系),这两种方式在原理、操作流程、应用场景上均有所区别,本文主要就瞬时转染与稳定转染之间的区别做一介绍。
- 0
- 0
- xiaoqin2023
- 发布于 2023-10-13 16:46
- 阅读 ( 3458 )