ggplot2点图geom_point中aes(shape)映射
此前在相关文章中详细介绍了关于geom_point 在设定shape时可以进行多少设置,每种出现的结果: http://www.omicsclass.com/article/475 区别与点shape设定,点的shape 映射必须转化成因子,连...
- 0
- 0
- Daitoue
- 发布于 2018-10-12 15:07
- 阅读 ( 8062 )

Cytoscape如何基于点的属性快速对点和相连的线进行颜色分类
在利用Cytoscape绘图的过程中如何对具有不同属性(分类)的点(以及与该点相连的线)进行颜色着色呢? 譬如下图 显示了miRNA 和靶基因的相互作用,并基于靶基因的类型对网网络图进行了颜色处理...
- 1
- 0
- Daitoue
- 发布于 2018-10-12 11:28
- 阅读 ( 21334 )
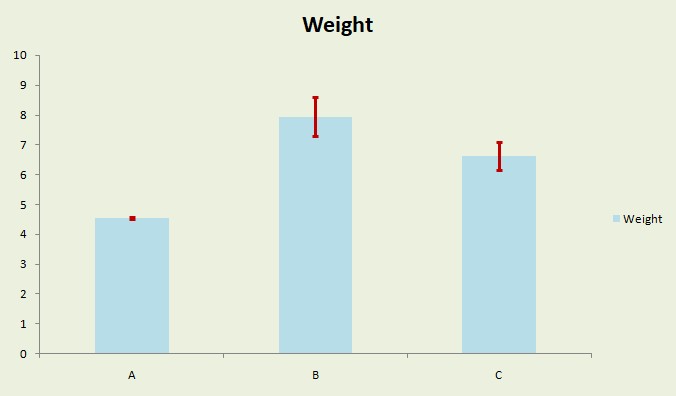
Excel画图过程中添加误差线
误差线的添加可以用标准差、标准误或者置信区间:http://www.omicsclass.com/article/474 那么在Excel中如何快速添加呢? 以下图中的测量数据的均值(Excel函数:AVERAGE计算)绘制出A、B、C...
- 0
- 0
- Daitoue
- 发布于 2018-10-11 17:13
- 阅读 ( 33145 )
网络概念以及Cytoscape导入数据格式理解(ceRNA等多元关系网络数据)
复杂网络(可参考维基百科Complex network、Network theory)由多个基本单元( 或节点) 与它们之间的相互作用( 即连线) 组成,典型的例子有互联网、神经网络和各类生物网络(包括代谢网络、基因调...
- 1
- 1
- Daitoue
- 发布于 2018-10-09 14:37
- 阅读 ( 13630 )
ggplot2点图geom_point中点的shape有多少?
ggplot2可以利用geom_point绘制散点图,而点的形状控制参数shape会显示多少效果呢?(注意此处只介绍shape的设定,不是aes(shape)映射) 可以通过查询?shape 获得以下内容: # Shape exampl...
- 0
- 1
- Daitoue
- 发布于 2018-09-30 16:59
- 阅读 ( 29444 )
误差线该用标准差还是标准误?
误差线对应的表示的到底是标准差还是标准误?其实……都可以,此外还可以用特定的置信区间(譬如,95%的区间) 误差线 主要指示数据每一个数据点的误差(或不确定性)范围,显示潜在的误差或相...
- 1
- 5
- Daitoue
- 发布于 2018-09-30 14:58
- 阅读 ( 46594 )
利用awk 对某一列数据取对数
awk 具有内置算术函数log(x),以e 为底可以对数据取对数运算,而以其他数值为底进行取对数可以基于这一函数进行转换,譬如 以2为底取对数: log(x)/log(2)即可。 下面针对数据第二列进行取对...
- 0
- 2
- Daitoue
- 发布于 2018-09-30 13:57
- 阅读 ( 13697 )