bedtools安装报错cc1plus: error: unrecognized command line option "-std=c++11"
bedtools 安装报错
- 0
- 0
- omicsgene
- 发布于 2018-09-29 10:56
- 阅读 ( 7332 )
ANNOVAR人类各个数据库变异注释结果表格说明
ANNOVAR人类各个数据库变异注释结果表格说明,SnpEff
- 3
- 10
- omicsgene
- 发布于 2018-09-28 20:06
- 阅读 ( 37741 )
1000G千人基因组人群分类亚群缩写对照
了解分类信息,有助于研究特定人群中突变的等位基因频率:Which populations are part of your study?
- 0
- 2
- omicsgene
- 发布于 2018-09-28 18:13
- 阅读 ( 17059 )

ggplot2控制坐标轴截距交点位置(原点)
ggplot2绘图结果往往X轴和Y轴的交点往往不是原点:譬如下图y轴就不是起始于0 调整坐标起始位点可以利用scale_y_continuous(expand = c(0, 0))或者scale_x_continuous(expand = c(0, 0)) expa...
- 0
- 0
- Daitoue
- 发布于 2018-09-21 17:05
- 阅读 ( 19444 )
ggplot2控制百分比坐标
在利用ggplot2绘图过程中常会要求Y轴显示为百分比类型,需要借助包scales 和ggplot2中的函数scale_y_continuous(labels)来控制 譬如绘制相关百分比或者密度频率等类型的图片时可以将对应的...
- 0
- 0
- Daitoue
- 发布于 2018-09-21 15:58
- 阅读 ( 11198 )
ggplot2-气泡图(控制点的大小范围)
Pathway富集结果常用气泡图来显示,其基本代码如下: 案例数据: > dat ko_id Kegg_pathway Rich_factor Pvalue DEGs1 ko00195 ...
- 1
- 2
- Daitoue
- 发布于 2018-09-21 15:04
- 阅读 ( 26066 )
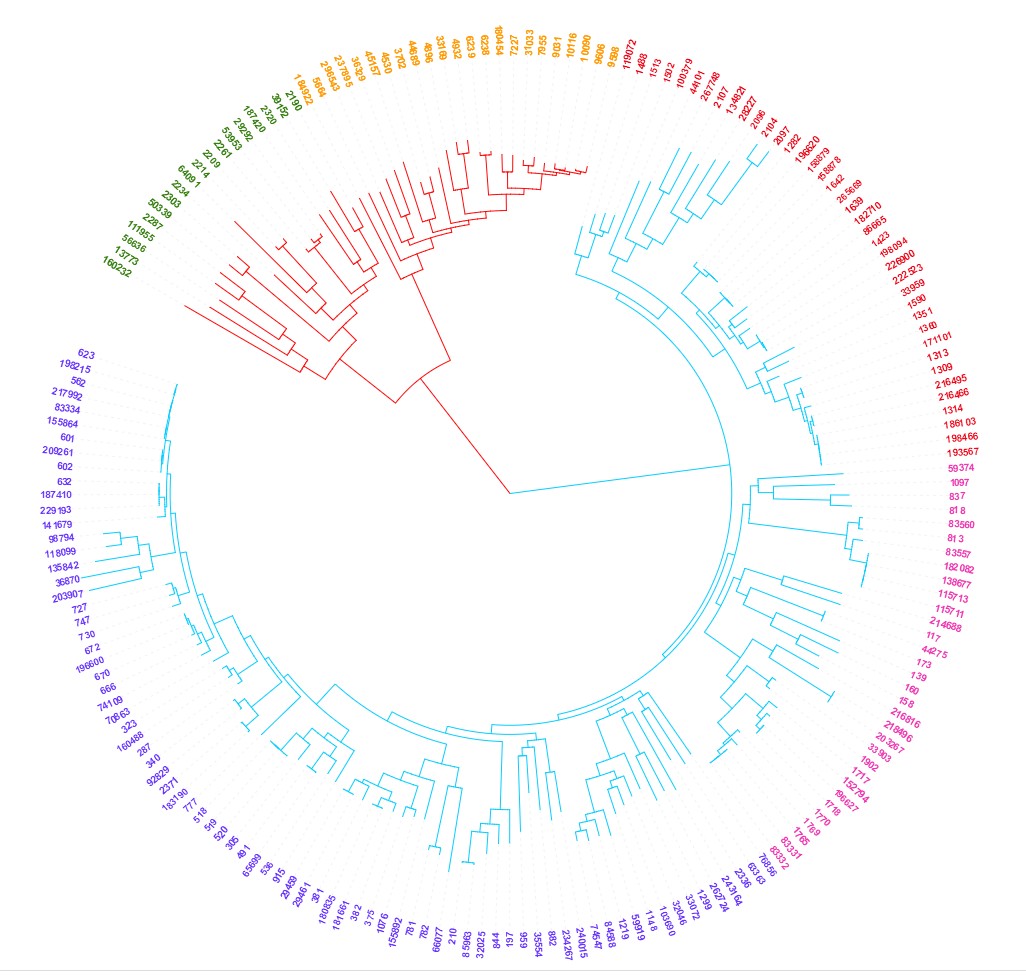
详细介绍如何利用iTOL在进化树中用颜色体现样品分组信息
详细介绍如何利用iTOL在进化树中用颜色体现样品分组信息。
- 3
- 2
- 安生水
- 发布于 2018-09-21 13:19
- 阅读 ( 35039 )
TCGA生存分析课程的数据准备
请问,在TCGA差异分析课程中的差异表达矩阵是用ensembleID表示lncrna,而TCGA生存分析的表达矩阵是用lncrna的gene symbol,请问是怎么转换的,用ensemble官网转换之后,代码总报错,请问该怎么...
- 0
- 0
- 盘子被夹子夹着
- 发布于 2018-09-20 15:00
- 阅读 ( 3704 )