ensembl和NCBI基因组下载,基因序列下载查看
参考基因组下载,Ensembl,NCBI,JGI等网站下载使用,可视化浏览,基因查看与下载等;
- 8
- 5
- omicsgene
- 发布于 2018-04-22 14:59
- 阅读 ( 52464 )

正选择分析之 Site Models
作为常用的正选择分析方法,计算Ka/Ks方法较为严格。本文介绍利用Sites Model方法来分析正选择基因。
- 3
- 5
- 安生水
- 发布于 2018-04-22 10:39
- 阅读 ( 17834 )
Orthofinder-构建物种系统发育树(Phylogenetic tree)
在前一篇文章里写了使用OthroFinder软件构建物种同源基因分析的工作原理的介绍,这次就利用Orthofinder结果中的数据来绘制物种系统发育树(Phylogenetic tree)。 在Orthofinder结果文件中有一...
- 0
- 4
- rzx
- 发布于 2023-06-15 17:33
- 阅读 ( 9973 )
进化树美化操作详解- iTOL
绘制进化树,是科研人的基础技能之一,而一张好看的进化树图,不仅能直观得展现基因或物种间的发育进化关系,更能增加文章的观赏性,赢得审稿人的青睐。今天给大家介绍的是一款非常实用且操作简...
- 2
- 4
- rzx
- 发布于 2022-10-12 15:53
- 阅读 ( 35405 )
送你一个小巧好用的序列分析软件-APE-功能强大,操作简单!
APE是一款序列分析软件,可以对序列进行酶切位点预测、序列比对、引物设计、DNA序列翻译等等,用途是十分广泛,APE软件小巧快速,操作简单,下面我们一起了解一下它的功能! 1. 酶切位点预测...
- 0
- 4
- 红橙子
- 发布于 2020-06-29 11:58
- 阅读 ( 20580 )
如何利用HMMER鉴定基因家族成员
作者 smyang2018 在可视化, 归档, 生物信息学, 软件学习 通常我们用的都是通过blast比对来确定我们需要的家族成员,首先是比对序列,再次是需要目标物种的蛋白序列,来进行比对,通常比对的时...
- 3
- 4
- smyang2018
- 发布于 2019-04-04 10:23
- 阅读 ( 15570 )
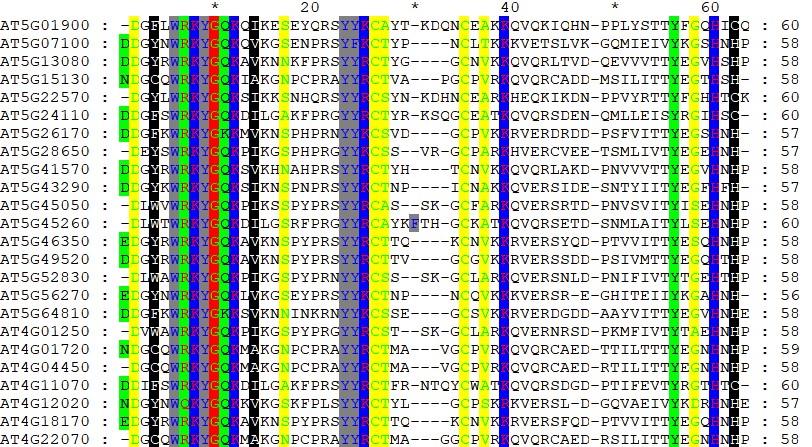
GeneDoc绘制漂亮的多重序列比对图
由于软件的功能限制,一些比对软件生成的比对文件,如Clustalx的*.aln文件,可读性差,无法满足高质量期刊的要求。因此,在实际操作过程中,需要对一些多重序列比对的文件进行着色美化。 GeneD...
- 6
- 4
- 安生水
- 发布于 2019-03-01 12:00
- 阅读 ( 26318 )
出众的进化树注释软件Evolview
Evolview是一款在线进化树注释软件,能够实现很复杂的注释效果,且用法并不复杂。
- 6
- 4
- 安生水
- 发布于 2018-11-28 09:20
- 阅读 ( 30173 )
blast比对结果说明
我们作比对时经常用到blast,其比对结果一般都用m8格式(即参数是 -m 8,blast+是 -outfmt 6),但是结果文件中是没有表头的,这里来写一下。
- 2
- 4
- 安生水
- 发布于 2018-07-20 11:13
- 阅读 ( 24531 )

你了解SAM和BAM文件吗
当我们测序得到的fastq文件map到基因组之后,会得到一个以SAM或Bam为扩展名的文件。这里将详细介绍SAM和Bam文件!
- 0
- 4
- 安生水
- 发布于 2018-06-29 09:33
- 阅读 ( 14952 )
如何按照影响因子筛选文章--PubMed文献搜索技巧
PubMed文献搜索的一个实用技能--如何按照影响因子进行筛选文章?
- 1
- 4
- 生信老顽童
- 发布于 2018-06-20 10:18
- 阅读 ( 24638 )
利用NCBI和Pfam数据库查找基因家族保守结构域相关信息
相近功能基因的检索方法,如何利用pfam数据库下载隐马尔科夫hmm文件。
- 6
- 4
- landy
- 发布于 2018-04-23 14:05
- 阅读 ( 60050 )
PASA报错:unique constraint failed: asmbl_link.asmbl_acc, asmbl_link.cdna_acc
通常会从同源、转录组、从头三个方面进行基因结构注释,其中PASA是转录组注释的常用软件。 报错: 把三代转录组数据投入PASA是出现报错:unique constraint failed: asmbl_link.asmbl_acc, as...
- 0
- 3
- Ti Amo
- 发布于 2024-09-30 13:50
- 阅读 ( 2140 )