python 版本的MCScanX分析物种之间分共线性
所有内容可与 基因家族分析 课程中学习!
MCscan可以用于查看多个基因组(或单个基因组内)中共线性区段的关系。今天我们介绍python版的MCscan(JCVI 包),其中包装了很多的绘图功能。这里介绍具体使用方法(以葡萄和桃子两基因组分析为例)。
官方网站:https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version)
1. 安装
这里可以分两步,分别是安装LASTAL和jcvi python包。
LASTAL下载地址: http://last.cbrc.jp/,编译后将 lastal 和 lastdb 添加到PATH中。
jcvi安装比较简单,在命令行直接运行下列命令:
pip install jcvi
2. 数据下载
运行MCscan需要序列以及坐标文件,分别是fasta格式以及BED格式的,例如,可以从Phytozome下载cds的序列文件以及gff文件,再由gff文件生成BED文件。当然JCVI有相应的方法,可以直接下载,省去我们自己下载整理数据的麻烦,具体操作如下:
$ python -m jcvi.apps.fetch phytozome
...
Acoerulea Alyrata Athaliana
Bdistachyon Brapa Cclementina
Cpapaya Creinhardtii Crubella
Csativus Csinensis Csubellipsoidea_C-169
Egrandis Fvesca Gmax
Graimondii Lusitatissimum Mdomestica
Mesculenta Mguttatus Mpusilla_CCMP1545
Mpusilla_RCC299 Mtruncatula Olucimarinus
Osativa Ppatens Ppersica
Ptrichocarpa Pvirgatum Pvulgaris
Rcommunis Sbicolor Sitalica
Slycopersicum Smoellendorffii Stuberosum
Tcacao Thalophila Vcarteri
Vvinifera Zmays early_release
...
$ python -m jcvi.apps.fetch phytozome Vvinifera,Ppersica
...
$ ls
Ppersica_139_cds.fa.gz Ppersica_139_gene.gff3.gz Vvinifera_145_cds.fa.gz Vvinifera_145_gene.gff3.gz
GFF转BED:
$ python -m jcvi.formats.gff bed --type=mRNA --key=Name Vvinifera_145_gene.gff3.gz -o grape.bed
$ python -m jcvi.formats.gff bed --type=mRNA --key=Name Ppersica_139_gene.gff3.gz -o peach.bed
如果参考基因组没有cds的序列文件,可下载染色体序列,根据gff文件提取cds序列。
3. 成对共线性搜索
准备好输入文件后,就可以进行共线性搜索了。首先要将工作目录切换到输入文件所在目录,然后运行如下命令:
$ python -m jcvi.compara.catalog ortholog grape peach 20:33:42 [base] lastdb peach peach.cds 20:34:13 [base] lastal -u 0 -P 64 -i3G -f BlastTab peach grape.cds >grape.peach.last 20:34:30 [synteny] Assuming --qbed=grape.bed --sbed=peach.bed 20:34:31 [blastfilter] Load BLAST file `grape.peach.last` (total 403868 lines) 20:34:31 [base] Load file `grape.peach.last` 20:34:38 [blastfilter] running the cscore filter (cscore>=0.70) .. 20:34:39 [blastfilter] after filter (294217->31229) .. 20:34:39 [blastfilter] running the local dups filter (tandem_Nmax=10) .. 20:34:39 [blastfilter] after filter (31229->21089) .. ... A total of 30654 (NR:18661) anchors found in 678 clusters. Stats: Min=4 Max=683 N=678 Mean=45.2123893805 SD=80.8407828815 Median=16.0 Sum=30654 NR stats: Min=4 Max=460 N=678 Mean=27.5235988201 SD=48.0461479616 Median=11.0 Sum=18661
运行结束后会得到以下几个文件
$ ls grape.peach.*
grape.peach.lifted.anchors grape.peach.anchors grape.peach.last.filtered grape.peach.last
4. 成对同线性可视化
观察成对的共线性的最佳可视化方法是使用dotplot,这里只需要一条命令。
$ python -m jcvi.graphics.dotplot grape.peach.anchors
生成一个 grape.peach.pdf 点图文件,如图:
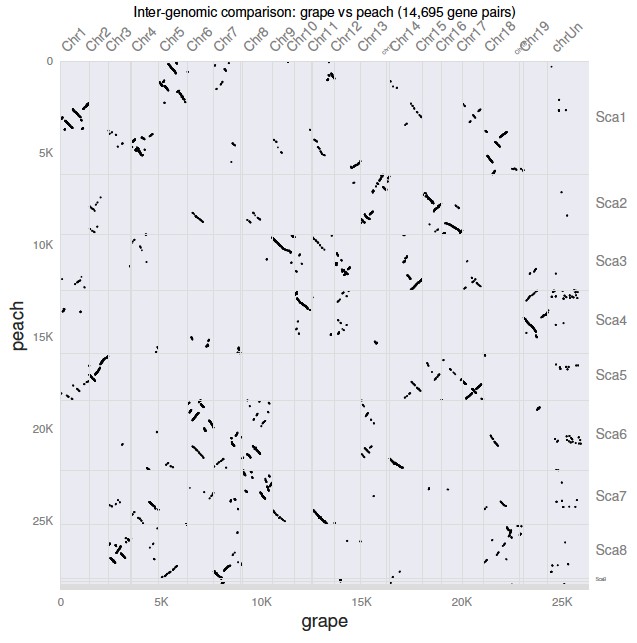
5. 线性可视化
我们还可以使用同样的共线性输出文件做不同的可视化图片。这里需要在准备两个输入文件,seqids 文件和 layout 文件。
前者为两个基因组都包括哪些的染色体,这里最好移除较短的scaffolds,文件格式如下所示:
chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19
scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8
其中第一行为葡萄所包含的19个染色体,第二行为桃子包含的8个染色体。
后者为画图所需的配置文件,格式如下:
# y, xstart, xend, rotation, color, label, va, bed
.6, .1, .8, 0, , Grape, top, grape.bed
.4, .1, .8, 0, , Peach, top, peach.bed
# edges
e, 0, 1, grape.peach.anchors.simple
其中前三列指定了轨迹的位置,然后是rotation, color, label, vertical alignment (va), 和 基因组 BED 文件.最后是连线的绘制,信息来自grape.peach.anchors.simple文件。
grape.peach.anchors.simple由以下命令生成:
$ python -m jcvi.compara.synteny screen --minspan=30 --simple grape.peach.anchors grape.peach.anchors.new
然后,我们就可以绘图了!
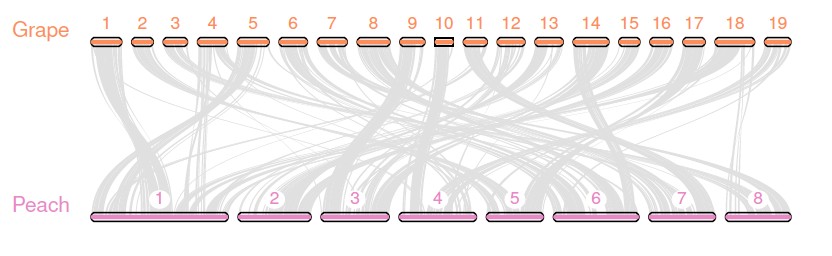
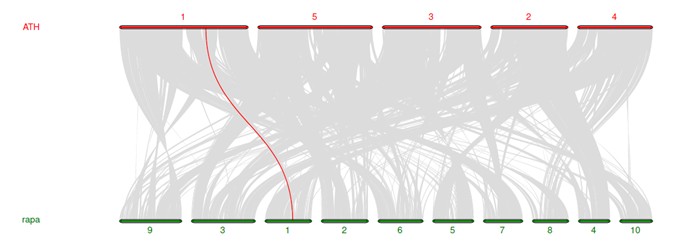
$ python -m jcvi.graphics.karyotype seqids layout
结果图看这里
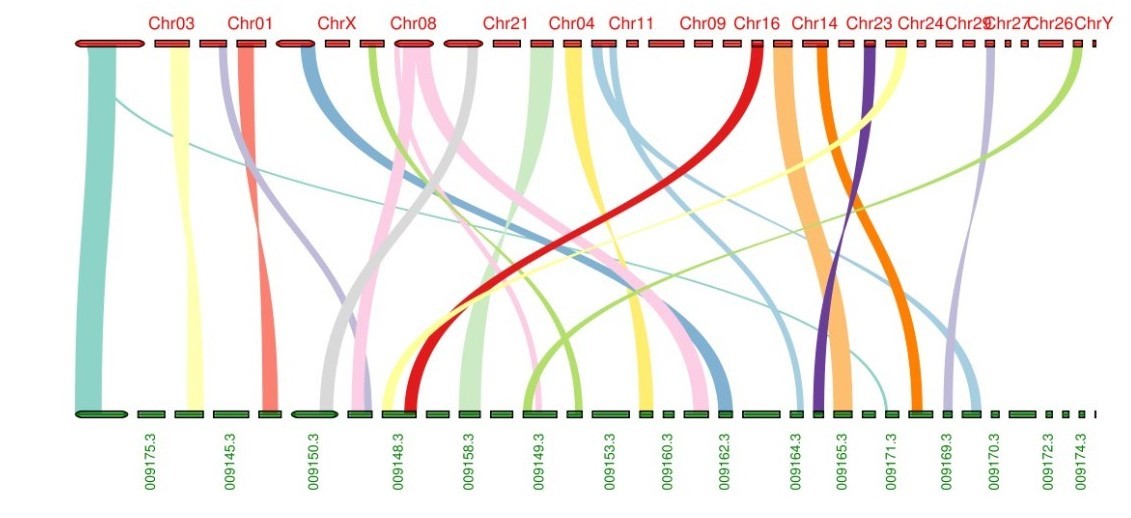
个性化设置显示颜色,最近做的一个基因组的共线性分析:
好了,到这里我们就结束了,想了解MCscan python版本的更多内容,可以查看MCscan 官网:https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version)
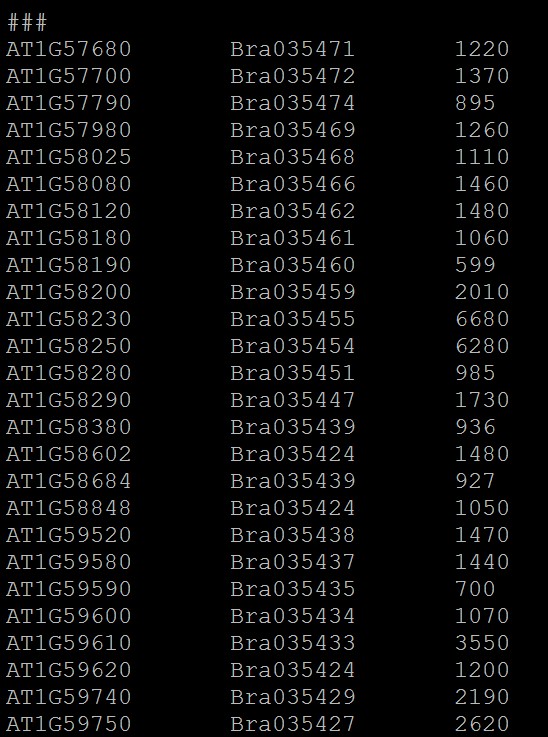
1.基因组共线性详细结果文件,过滤掉不可靠的共线性区块之后的文件:*.anchors.new,第一列为一个 基因组基因ID,第二列为另一个基因组的基因ID文件,即是两个基因组共线性区域内基因对应关系;
不同的共线性区块用“###” 隔开
2.基因组共线性简化结果文件:* anchors.simple;一行为一个共线性区块,前两列表示一个基因组中两个基因组之间的区域,与后两列(3,4列)另一个基因组中的两个基因之间的区域存在共线性关系;
第5列共线性打分值,最后一列为: +为正向,-为反向
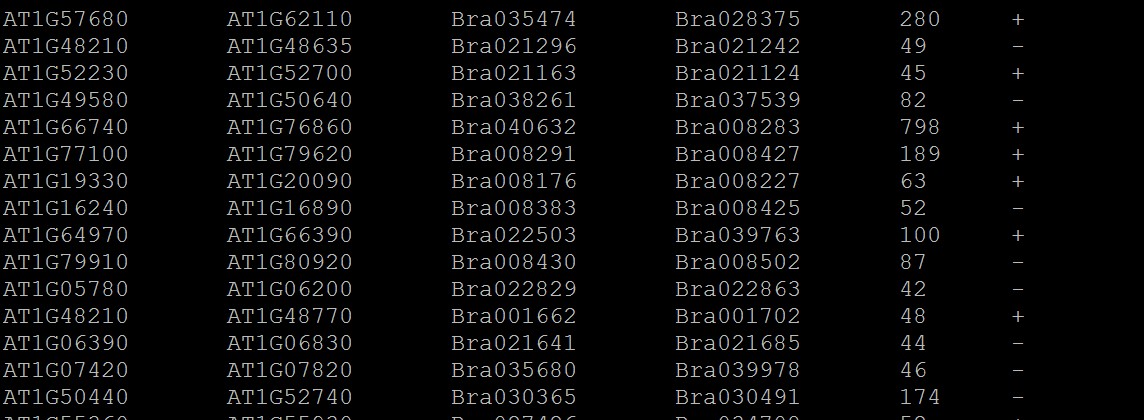
3.如果要设置颜色,可修改2.中的文件,也就是在需要修改颜色的行头部加上颜色值,具体格式如下:
注意颜色值为16进制,*隔开; 没有添加颜色值的行默认灰色:
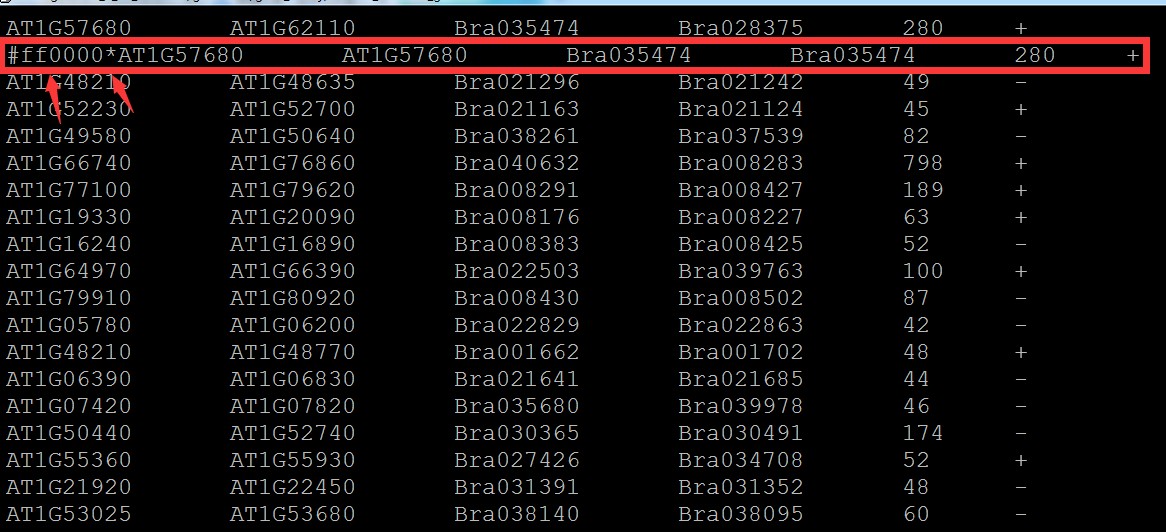
添加一行后:
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
- 发表于 2018-07-12 17:14
- 阅读 ( 31783 )
- 分类:软件工具
