TCGA临床数据下载—TCGAbiolinks
前面我们介绍了TCGAbiolinks下载数据,这里详细介绍一下如何下载临床数据:
TCGA中三种临床数据类型:

TCGAbiolinks中临床数据有以下三种类型,以及他们的区别如下:
In GDC database the clinical data can be retrieved from different sources:
- indexed clinical: a refined clinical data that is created using the XML files.
- XML files: original source of the data
- BCR Biotab: tsv files parsed from XML files
There are two main differences between the indexed clinical and XML files:
- XML has more information: radiation, drugs information, follow-ups, biospecimen, etc. So the indexed one is only a subset of the XML files
- The indexed data contains the updated data with the follow up information. For example: if the patient is alive in the first time clinical data was collect and the in the next follow-up he is dead, the indexed data will show dead. The XML will have two fields, one for the first time saying he is alive (in the clinical part) and the follow-up saying he is dead.
官方使用说明地址:http://bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/clinical.html
1.BCR Biotab(表格数据)
In this example we will fetch clinical data from BCR Biotab files.
query <-GDCquery(project = "TCGA-ACC", data.category = "Clinical", data.type = "Clinical Supplement", data.format = "BCR Biotab") GDCdownload(query) clinical.BCRtab.all <-GDCprepare(query) names(clinical.BCRtab.all) query <-GDCquery(project = "TCGA-ACC", data.category = "Clinical", data.type = "Clinical Supplement", data.format = "BCR Biotab", file.type = "radiation") GDCdownload(query) clinical.BCRtab.radiation <-GDCprepare(query)
2.Clinical indexed data(简化版本临床数据)
In this example we will fetch clinical indexed data (same as showed in the data portal).
clinical <-GDCquery_clinic(project = "TCGA-STAD", type = "clinical")
write.csv(clinical, file = 'TCGA-STAD_Clinical.csv', row.names =F)
这种临床数据已经很全了,但是对于有些癌种,有些临床数据是没有的,比如Gleason分级数据,是一种被广泛采用的前列腺癌组织学分级的方法,这种下载方法里面没有这种数据;
因此最全的临床数据下载方法是下面的第三种方法:
3.XML clinical data (推荐使用 信息最全)
The process to get data directly from the XML are: 1. Use GDCquery and GDCDownload functions to search/download either biospecimen or clinical XML files 2. Use GDCprepare_clinic function to parse the XML files.
The relation between one patient and other clinical information are 1:n, one patient could have several radiation treatments. For that reason, we only give the option to parse individual tables (only drug information, only radiation informtaion,…) The selection of the tabel is done by the argument clinical.info.
| Clinical | drug 用药信息 |
| Clinical | admin |
| Clinical | follow_up 患者最近一次随访/最终的生存数据 |
| Clinical | radiation 放疗信息 |
| Clinical | patient 收录患者大部分的临床信息 |
| Clinical | stage_event |
| Clinical | new_tumor_event 复发/转移 等信息 |
| Biospecimen | sample |
| Biospecimen | bio_patient |
| Biospecimen | analyte |
| Biospecimen | aliquot |
| Biospecimen | protocol |
| Biospecimen | portion |
| Biospecimen | slide |
| Other | msi |
Below are several examples fetching clinical data directly from the clinical XML files.
query <-GDCquery(project = "TCGA-STAD",
data.category = "Clinical", file.type = "xml") GDCdownload(query) clinical <-GDCprepare_clinic(query, clinical.info = "patient") #上表中黄色部分可设置 #循环输出所有的临床数据: clinical.info<-c("drug","follow_up","radiation","patient","stage_event","new_tumor_event","admin") for(i in clinical.info){ clinical <- GDCprepare_clinic(query, clinical.info = i) write.csv(clinical, file = paste0('TCGA-STAD_clinical_',i,'.csv'), row.names =F) }
4.数据合并问题 (第三种方法)
这几个矩阵c("drug","follow_up","radiation","patient","stage_event","new_tumor_event","admin")可不可以直接并成一个矩阵呢?
答案是:不可以合并多个表格的数据。
这可能是因为:
1. 随访曾中断过: 存在多次随访
2. 出现了新的癌症:不同的癌症记录,随访时间不一样
3. 接受手术治疗: 接受了手术的治疗,增加了新的随访数据
举个例子,下面这个患者,其在 clinical.patient (一般是第一次随访记录)和 clinical.patient.followup (会有多次随访记录)中的信息如下:
所以clinical.patient.followup 是最终的生存数据:
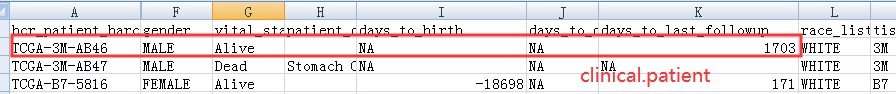
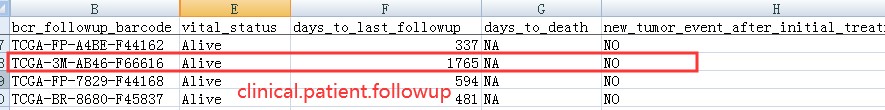
多次随访数据:

另外注意的是:Clinical indexed data 方法下载的数据中的:days_to_last_followup 是最后一次的随访数据。
再例如,clinical.patient 中某个患者其 has_new_tumor_events_information 列的值为 YES,则表示其在 clinical.new_tumor_event 中是有信息的,否则表示 clinical.new_tumor_event 中并无该患者的数据(也存在意外,小编就看到has_radiations_information == "NO", 但是却在clinical.radiation中有信息的患者),总之,几个矩阵并不是行数一致的!
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、biolinux搭建生物信息分析环境、linux命令处理生物大数据、perl入门到精通、perl语言高级、R语言画图、R语言快速入门与提高、python语言入门到精通
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、 GEO芯片数据不同平台标准化 、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代fastq测序数据解读、
9.全部课程可点击:组学大讲堂视频课程
- 发表于 2019-11-29 13:45
- 阅读 ( 15583 )
- 分类:TCGA
