TCGA数据库重大更新-数据如何下载?
TCGA数据库重大更新-数据如何下载?
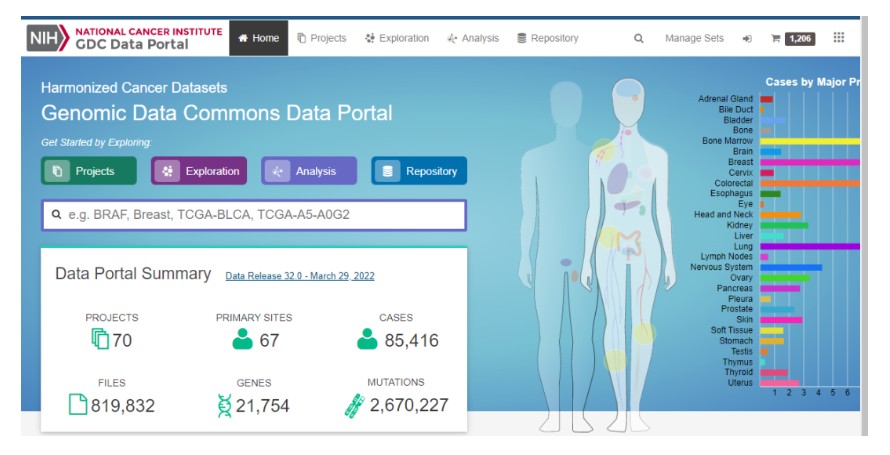
最近有粉丝留言,TCGA数据库发生更新,下载数据时报错。详细的更新信息参考TCGA官网说明:
https://docs.gdc.cancer.gov/Data/Release_Notes/Data_Release_Notes/#data-release-320
这里提示几点重要更新:
例如:转录组之前HTSeq流程的数据,现在是STAR-Counts的数据。
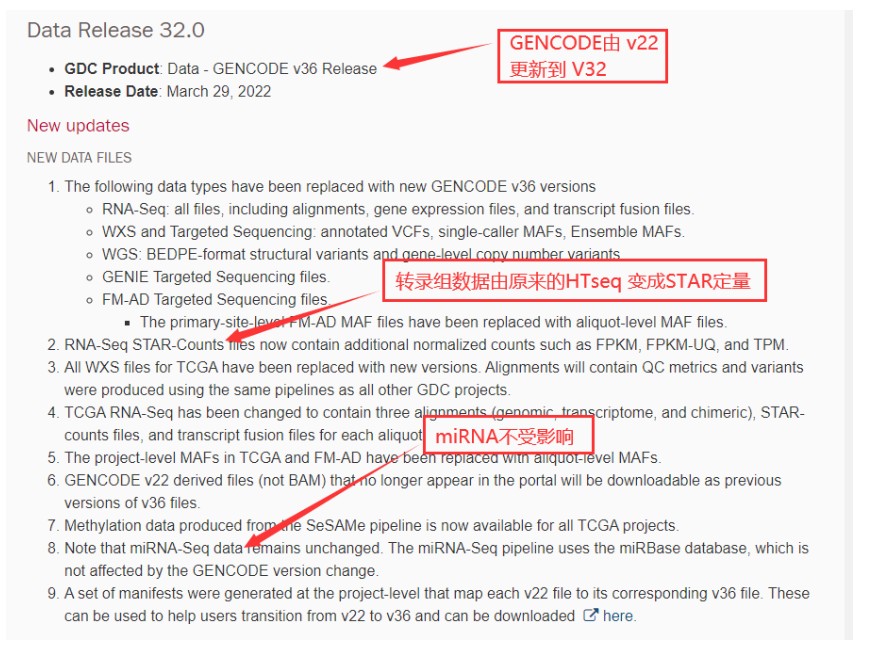
受到影响的主要是表达数据,这里大家可以更新一下TCGA下载的代码,即可下载最新的数据,详细代码及说明如下:
#安装最新的 TCGAbiolinks R 包
BiocManager::install("BioinformaticsFMRP/TCGAbiolinks")
#一个方法,可以将ensembl ID 转换成 gene name
ensemblToName<-function(expr,gene.info){
expr=as.data.frame(expr)
#顺序保持一致
gene.info<-as.data.frame(gene.info)
row.names(gene.info)<-gene.info$gene_id
gene.info<-gene.info[rownames(expr),]
sample.id=colnames(expr)
#添加gene name列 相同基因随机选取
gene_name_rmdup=!duplicated(gene.info$gene_name)
expr.res=expr[gene_name_rmdup,]
gene.info=gene.info[gene_name_rmdup,]
rownames(expr.res)<-gene.info$gene_name
expr.res
}
#加载需要的包,这里需要两个R包
package_list <- c("SummarizedExperiment","TCGAbiolinks")
for(p in package_list){
if(!suppressWarnings(suppressMessages(require(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))){
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(p)
suppressWarnings(suppressMessages(library(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))
}
}
#查询TCGA数据,这里以TCGA-CHOL为例,样本少好下载
query <- GDCquery(project = "TCGA-CHOL",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification", # 有些样本只有 "Isoform Expression Quantification"
workflow.type = "STAR - Counts",
#sample.type = sample_type,
legacy = FALSE)
files <- getResults(query,cols=c("cases"))
cat("Total files to download:", length(files))
# TP 样本数量 肿瘤样本
dataSmTP <- TCGAquery_SampleTypes(barcode = files, typesample = "TP")
cat("Total TP samples to down:", length(dataSmTP))
# NT 样本数量 正常组织样本
dataSmNT <- TCGAquery_SampleTypes(barcode = files,typesample = "NT")
cat("Total NT samples to down:", length(dataSmNT))
#下载数据
GDCdownload(query = query,directory = "TCGA-CHOL",files.per.chunk=10, method='api')
# 保存整理下载数据结果
gene.data <- GDCprepare(query = query,directory = "TCGA-CHOL" )
#获得样本数据信息
sample.info.list=colData(gene.data)@listData
sample.info=as.data.frame(sample.info.list[1:10])
#获得基因信息数据
gene.info=as.data.frame(rowRanges(gene.data))
# 获得表达数据 ,注意这里表达数据有6种,可以根据i参数指定:
# unstranded stranded_first stranded_second tpm_unstrand fpkm_unstrand fpkm_uq_unstrand
data_expr <- assay(gene.data,i = "unstranded")
#修改ensembl ID 为基因name
data_expr<-ensemblToName(data_expr,gene.info)
#数据写出
write.table(data.frame(ID=rownames(data_expr),data_expr,check.names = F), file = 'gene_expression_Counts.tsv', sep="\t",row.names =F, quote = F)
write.table(data.frame(ID=rownames(gene.info),gene.info,check.names = F), file = 'gene.info.tsv', sep="\t",row.names =F, quote = F)
write.table(data.frame(ID=rownames(sample.info),sample.info,check.names = F), file = 'sample.info.tsv', sep="\t",row.names =F, quote = F)
生信博士提供全自动数据下载脚本:
命令行运行:
为保证代码正常运行成功,确保安装最新的R包 TCGAbiolinks,安装方法:
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("BioinformaticsFMRP/TCGAbiolinksGUI.data") BiocManager::install("BioinformaticsFMRP/TCGAbiolinks")
或者用我们组学大讲堂提供的最新 docker镜像,里面已经更新了最新的TCGAbiolinks包:
omicsclass/r-server:lastest
用命令行代码《tcga_gene_exp_download.r》自动下载表达数据,运行命令如下:
Rscript tcga_gene_exp_download.r -p TCGA-CHOL
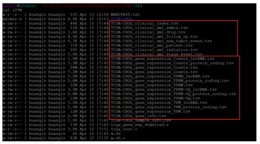
结果展示:
运行完上面的命令行,即可自动下载临床数据和表达数据,并自动整理成表格,方便后续分析:
如何使用命令行的方法分析数据
扫描下方二维码学习命令行分析数据,以及获得TCGA数据下载最新代码:
https://zzw.h5.xeknow.com/s/3mdwk4

- 发表于 2022-04-28 17:48
- 阅读 ( 8780 )
- 分类:TCGA
