monocle3手动选取细胞发育轨迹分析
monocle3手动选取细胞发育轨迹分析
接之前分析:
monocle3相对于monocle2的升级我觉得最重要的就是可以手动在umap图赏选取起始细胞和终止细胞分析特定轨迹,这样可以个性化的分析自己关注的细胞亚群发育轨迹及关键表达变化的基因。
library(qs)
library(scop)
library(monocle3)
# 设置工作路径
setwd("~/maize_root/07.monocle3")
cds <- load_monocle_objects(directory_path = "monocle.Columella2//monocle3_cds.data/")
obj <- qread("../06.subset/Columella.added.subtype.qs") # 读入seurat对象
# 将cds UMAP替换seurat obj里面的umap
# 如果你跑monocle3是用的seurat降维结果(--use.seurat.umap),这步可以省略
umap_embeddings <- reducedDims(cds)$UMAP
colnames(umap_embeddings) <- c("umap_1", "umap_2")
obj[["umap"]]@cell.embeddings <- umap_embeddings[colnames(obj), ]
# 可视化结果
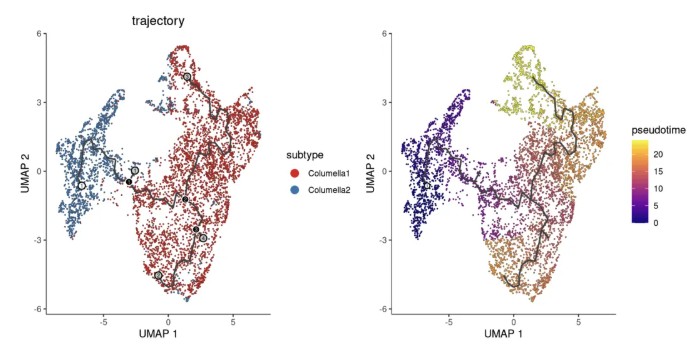
p1 <- plot_cells(cds,
color_cells_by = "subtype",
label_cell_groups = F,
label_groups_by_cluster = F,
label_leaves = T,
label_branch_points = TRUE,
reduction_method = "UMAP"
) +
ggtitle("trajectory") +
scale_color_manual(values = color_palette) +
theme(plot.title = element_text(hjust = 0.5))
p2 <- plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups = FALSE,
label_leaves = FALSE,
label_branch_points = FALSE,
graph_label_size = 1.5
)
p1 + p2
轨迹拐点ID显示:graph_label_size 点大小,label_principal_points 显示主点.
plot_cells(cds, color_cells_by = "pseudotime", label_cell_groups = FALSE, label_leaves = FALSE, label_branch_points = FALSE, label_principal_points = TRUE, group_label_size = 2, labels_per_group = 1, graph_label_size = 5 )
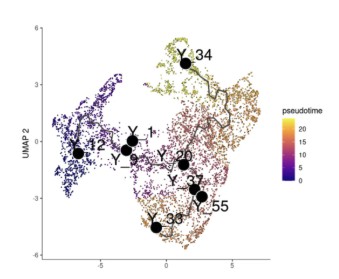
以Columella2 细胞为发育起点细胞,很明显有两个发育方向。根据节点名称选取感兴趣的两个细胞发育轨迹:
Y_12 -> Y_33
Y_12 -> Y_34
obj$Monocle3_Pseudotime <- pseudotime(cds)[colnames(obj)]
# Select the lineage using monocle3::choose_graph_segments
cds_sub <- choose_graph_segments(
cds,
starting_pr_node = "Y_12",
ending_pr_nodes = "Y_33"
)
obj$Lineage_1 <- NA
obj$Lineage_1[colnames(cds_sub)] <- obj$Monocle3_Pseudotime[colnames(cds_sub)]
CellDimPlot(
obj,
group.by = "subtype",
lineages = "Lineage_1",
lineages_span = 0.3,
theme_use = "theme_blank"
)
# 第二条发育轨迹
cds_sub <- choose_graph_segments(
cds,
starting_pr_node = "Y_12",
ending_pr_nodes = "Y_34"
)
obj$Lineage_2 <- NA
obj$Lineage_2[colnames(cds_sub)] <- obj$Monocle3_Pseudotime[colnames(cds_sub)]
CellDimPlot(
obj,
group.by = "subtype",
lineages = "Lineage_2",
lineages_span = 0.2,
theme_use = "theme_blank"
)
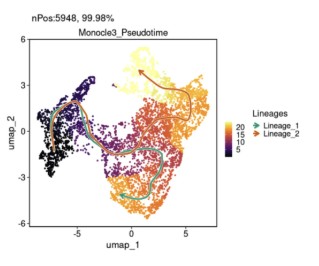
# 合并绘图
FeatureDimPlot(
obj,
features = "Monocle3_Pseudotime",
reduction = "UMAP",
lineages_span = 0.3,
lineages = paste0("Lineage_", 1:2),
palette = "inferno"
)
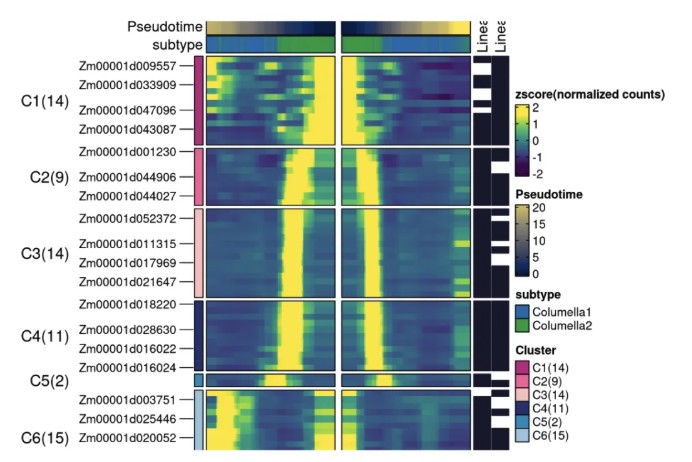
发育轨迹表达变化基因展示:
obj <- RunDynamicFeatures(
srt = obj,
lineages = c("Lineage_1", "Lineage_2"),
n_candidates = 200
)
ht <- DynamicHeatmap(
srt = obj,
lineages = c("Lineage_1", "Lineage_2"),
use_fitted = TRUE,
n_split = 6,
reverse_ht = "Lineage_1",
heatmap_palette = "viridis",
cell_annotation = "subtype",
pseudotime_label = 25,
pseudotime_label_color = "red",
height = 5,
width = 1.5
)
print(ht$plot)
如果遇到软件安装错误无法运行,可以使用组学大讲堂云服务器,已经安装好所有单细胞转录组数据分析的软件运行环境,省去环境配置麻烦,详情可扫下方二维码咨询:

Wang, T., Wang, F., Deng, S. et al. Single-cell transcriptomes reveal spatiotemporal heat stress response in maize roots. Nat Commun16, 177 (2025). https://doi.org/10.1038/s41467-024-55485-3
跟多生信文章免费视频请关注,生信课堂:

- 发表于 2026-04-14 08:54
- 阅读 ( 661 )
- 分类:转录组
