单细胞转录组拟时序-monocle3分析代码
接之前分析:1.玉米根热胁迫单细胞转录组复现2.玉米根单细胞转录组细胞类型注释代码3.热胁迫处理玉米根单细胞转录组差异基因分析
Monocle3于2020年正式发表,相比2017年发表于《Nature Methods》的Monocle2,在算法架构、功能整合及数据处理能力上实现了系统性升级。其中最显著的升级之一是轨迹推断的灵活性:Monocle3采用了基于UMAP的降维与图学习框架,能够重建包含多分支、树状甚至环路结构的复杂轨迹,而Monocle2主要处理树状结构。另一个关键升级在于手动选择轨迹起始点的功能——Monocle3支持用户直接在降维图上交互式地选择一个或多个节点作为轨迹的“根”,或通过细胞注释信息让算法自动推断,从而更精确地控制伪时间分析的生物学起点;而Monocle2在这方面的灵活性较低。
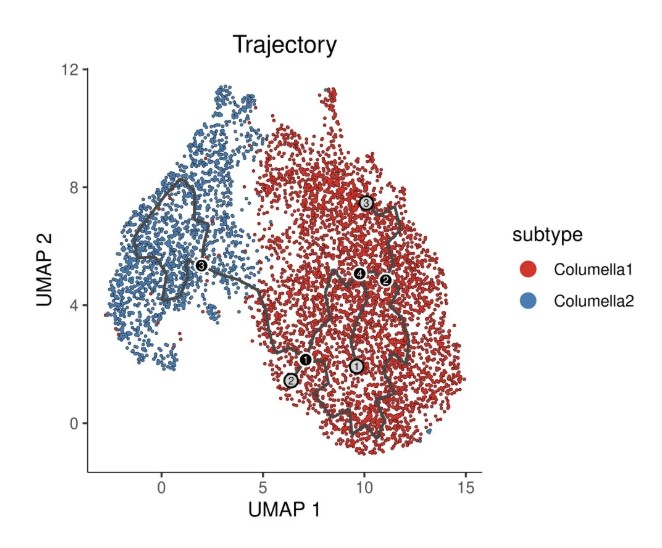
下图为玉米根Columella细胞再聚类分成两类,结合CytoTrace分析Columella2分化潜力更高,即CytoTrace打分更高,我们将其设为起始细胞,利用monocle3做轨迹分析:
Monocle3 单细胞轨迹分析代码
1. 从 Seurat 对象构建 Monocle3 CDS 对象
2. 导入 Seurat 预计算的 UMAP 降维结果
3. 细胞聚类与分区(partition)识别
4. 轨迹图学习与构建
5. 拟时序分析(以 Columella2 为起点)
6. 拟时序差异基因鉴定(Moran's I 检验)
7. 基因共表达模块分析
8. 基因表达趋势聚类与功能富集分析
加载依赖包
suppressPackageStartupMessages({
library(Seurat)
library(monocle3)
library(SeuratWrappers)
library(ggplot2)
library(dplyr)
library(pheatmap)
library(ClusterGVis)
})
1. 数据读取与CDS对象构建
setwd("~/maize_root/07.monocle3")
# 读取 Seurat 对象(玉米根尖 Columella 细胞亚群)
# 使用 qs 格式实现快速读写,多线程加速
seurat_obj <- qread("../06.subset/Columella.added.subtype.qs", nthreads = 4)
# 自定义颜色向量(73种高区分度颜色,用于分组着色)
dimCols <- c(
"#E41A1C", "#377EB8", "#4DAF4A", "#984EA3", "#FF7F00", "#A65628",
"#F781BF", "#999999", "#66C2A5", "#FC8D62", "#8DA0CB", "#E78AC3",
"#A6D854", "#FFD92F", "#E5C494", "#B3B3B3", "#7FC97F", "#BEAED4",
"#FDC086", "#386CB0", "#F0027F", "#BF5B17", "#666666", "#1B9E77",
"#D95F02", "#7570B3", "#E7298A", "#66A61E", "#E6AB02", "#A6761D",
"#A6CEE3", "#1F78B4", "#B2DF8A", "#33A02C", "#FB9A99", "#E31A1C",
"#FDBF6F", "#CAB2D6", "#6A3D9A", "#FFFF99", "#B15928", "#8DD3C7",
"#FFFFB3", "#BEBADA", "#FB8072", "#80B1D3", "#FDB462", "#B3DE69",
"#FCCDE5", "#D9D9D9", "#BC80BD", "#CCEBC5", "#FFED6F", "#FBB4AE",
"#B3CDE3", "#DECBE4", "#FED9A6", "#FFFFCC", "#E5D8BD", "#FDDAEC",
"#F2F2F2", "#B3E2CD", "#FDCDAC", "#CBD5E8", "#F4CAE4", "#E6F5C9",
"#FFF2AE", "#F1E2CC", "#CCCCCC"
)
# 提取单细胞表达矩阵(原始 counts 数据,UMI 计数)
expression_matrix <- LayerData(seurat_obj, assay = "RNA", layer = "counts")
# 提取细胞元数据(分组信息、样本来源等)
cell_metadata <- seurat_obj@meta.data
# 构建基因注释数据框(Monocle3 必需)
gene_annotation <- data.frame(gene_short_name = rownames(expression_matrix))
rownames(gene_annotation) <- rownames(expression_matrix)
# 创建 Monocle3 的 CDS(Cell Data Set)对象
cds <- new_cell_data_set(
expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation
)
2. 降维可视化(导入 Seurat 预计算结果)
# 导入 Seurat 的 PCA 降维结果
cds@int_colData$reducedDims$PCA <- seurat_obj@reductions$pca@cell.embeddings
# 导入 Seurat 的 UMAP 降维结果
int.embed <- Embeddings(seurat_obj, reduction = "umap")
cds@int_colData$reducedDims$UMAP <- int.embed
# 导入 Seurat 的 t-SNE 降维结果(备用)
int.embed <- Embeddings(seurat_obj, reduction = "tsne")
cds@int_colData$reducedDims$tSNE <- int.embed
# 预处理:计算 PCA,指定维度数为 50
cds <- preprocess_cds(cds, num_dim = 50)
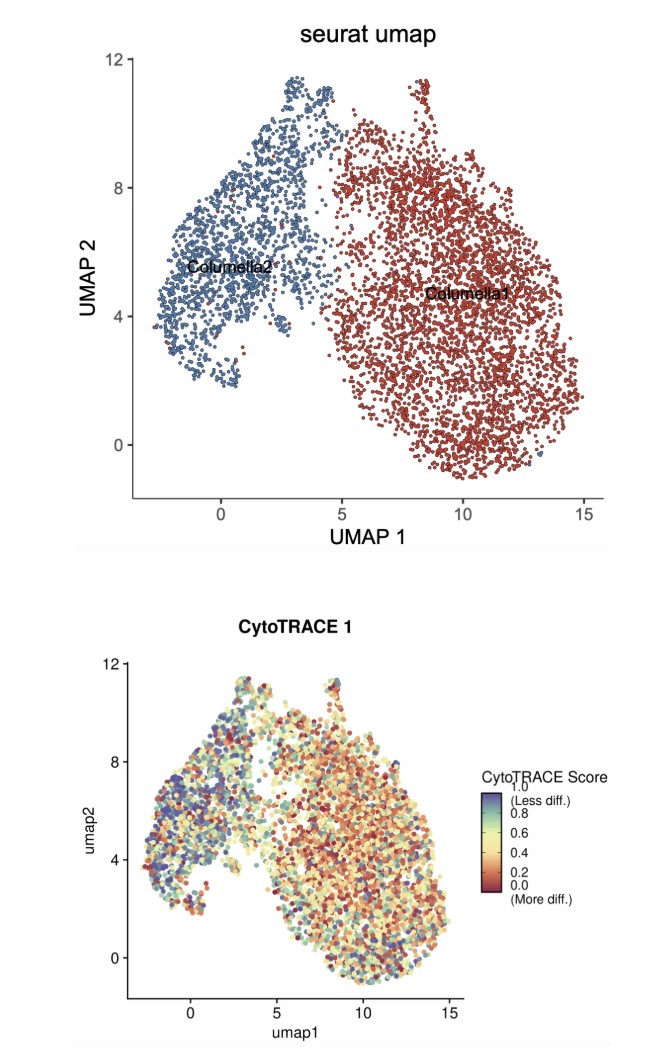
# 绘制 Seurat UMAP 图(按 subtype 分组着色)
plot_cells(cds,
reduction_method = "UMAP",
group_label_size = 3,
color_cells_by = "subtype",
show_trajectory_graph = FALSE,
cell_size = 0.4
) +
ggtitle("seurat umap") +
scale_color_manual(values = dimCols) +
theme(plot.title = element_text(hjust = 0.5))
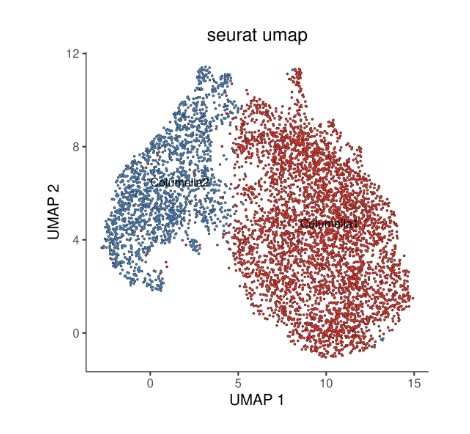
3. 细胞聚类与分区识别
# 聚类参数说明:
# - k = 20: k-nearest neighbors 数量
# - cluster_method = "leiden": 使用 Leiden 算法(优于 Louvain)
# - partition_qval = 0.05: 分区显著性阈值
cds <- cluster_cells(cds,
reduction_method = "UMAP",
k = 20,
cluster_method = "leiden",
partition_qval = 0.05
)
# 输出分区数量(Monocle3 自动识别轨迹分支数)
cat("Number of partitions:",
length(unique(partitions(cds, reduction_method = "UMAP"))), "\n")
# 绘制分区图(不同颜色代表不同分区)
plot_cells(cds,
show_trajectory_graph = FALSE,
group_label_size = 3,
cell_size = 0.4,
color_cells_by = "partition",
reduction_method = "UMAP"
) +
ggtitle("partition") +
theme(plot.title = element_text(hjust = 0.5))
这里所有的细胞划分到一个partition中,如果有多个partition根据不同的起始细胞设置分析不同的partition。
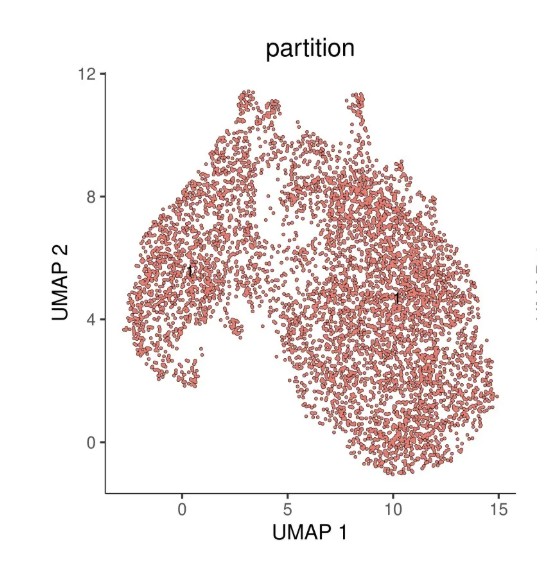
4. 轨迹图学习与构建
message("learn_graph start ...")
# 学习轨迹图结构(构建细胞状态转换的主图)
# learn_graph_control = NULL 使用默认参数
cds <- learn_graph(cds, learn_graph_control = NULL)
# 绘制轨迹图(按 subtype 着色,显示分支点和末端)
plot_cells(cds,
color_cells_by = "subtype",
group_label_size = 3,
cell_size = 0.4,
label_cell_groups = FALSE,
label_groups_by_cluster = FALSE,
label_leaves = TRUE, # 显示末端节点(灰色圆圈数字)
label_branch_points = TRUE, # 显示分支点(黑色圆圈数字)
reduction_method = "UMAP"
) +
ggtitle("Trajectory") +
scale_color_manual(values = dimCols) +
theme(plot.title = element_text(hjust = 0.5))
# 保存分区信息到 cell metadata
colData(cds)["partition"] <- cds@clusters$UMAP$partitions
pn <- length(unique(cds@clusters$UMAP$partitions))
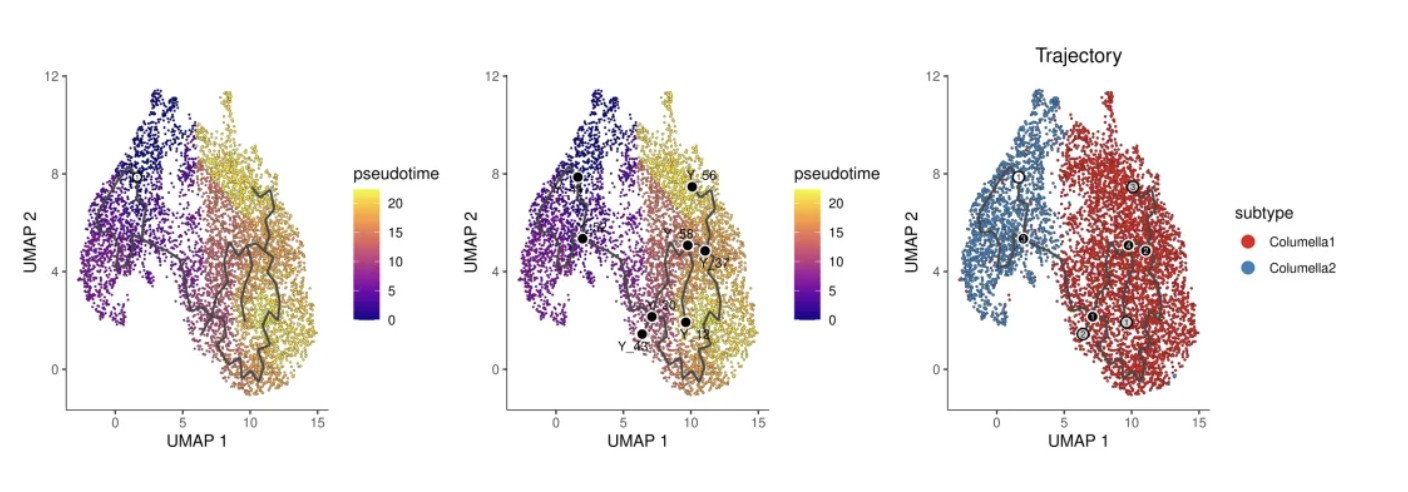
注:黑色的线显示的是graph的结构。数字带灰色圆圈表示不同的结局,也就是叶子。数字带黑色圆圈代表分叉点,从这个点开始,细胞可以有多个结局。这些数字可以通过label_leaves和label_branch_points参数设置。
5. 拟时序分析(指定根细胞)
# 根细胞设定:Columella2(柱状细胞亚型2,作为发育起点)
root_cell_type <- "Columella2"
# 找到属于根细胞类型的细胞索引
cell_ids <- which(cds@colData[, "subtype"] == root_cell_type)
# 获取每个细胞在 UMAP 主图中最近邻的顶点
closest_vertex <- cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertex
closest_vertex <- as.matrix(closest_vertex[colnames(cds), ])
# 确定根细胞对应的主图节点(取出现频率最高的节点)
root_pr_nodes <- igraph::V(principal_graph(cds)[["UMAP"]])$name[
as.numeric(names(which.max(table(closest_vertex[cell_ids, ]))))
]
# 按根节点排序细胞,计算拟时序值
cds <- order_cells(cds, root_pr_nodes = root_pr_nodes)
# 绘制拟时序 UMAP 图(颜色表示分化程度,黄色=早期,紫色=晚期)
plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups = FALSE,
label_leaves = FALSE,
label_branch_points = FALSE,
graph_label_size = 1.5,
show_trajectory_graph = FALSE
)
# 绘制带轨迹线的拟时序图(显示主图结构)
plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups = FALSE,
label_leaves = FALSE,
label_branch_points = FALSE,
graph_label_size = 1.5,
show_trajectory_graph = TRUE
)
# 绘制带主图节点标注的拟时序图(显示节点标签)
plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups = FALSE,
label_leaves = FALSE,
label_branch_points = FALSE,
label_principal_points = TRUE, # 显示主图节点
group_label_size = 2,
labels_per_group = 1,
graph_label_size = 2
)
# 绘制轨迹图(按 subtype 着色,带分支点和末端标注)
plot_cells(cds,
color_cells_by = "subtype",
group_label_size = 3,
cell_size = 0.4,
label_cell_groups = FALSE,
label_groups_by_cluster = FALSE,
label_leaves = TRUE,
label_branch_points = TRUE,
reduction_method = "UMAP"
) +
ggtitle("Trajectory") +
scale_color_manual(values = dimCols) +
theme(plot.title = element_text(hjust = 0.5))
6. 拟时序差异基因分析(Moran's I 检验)
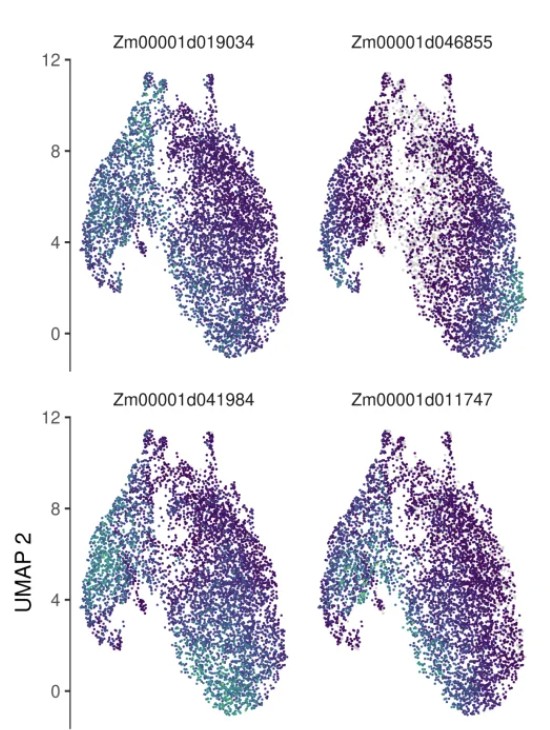
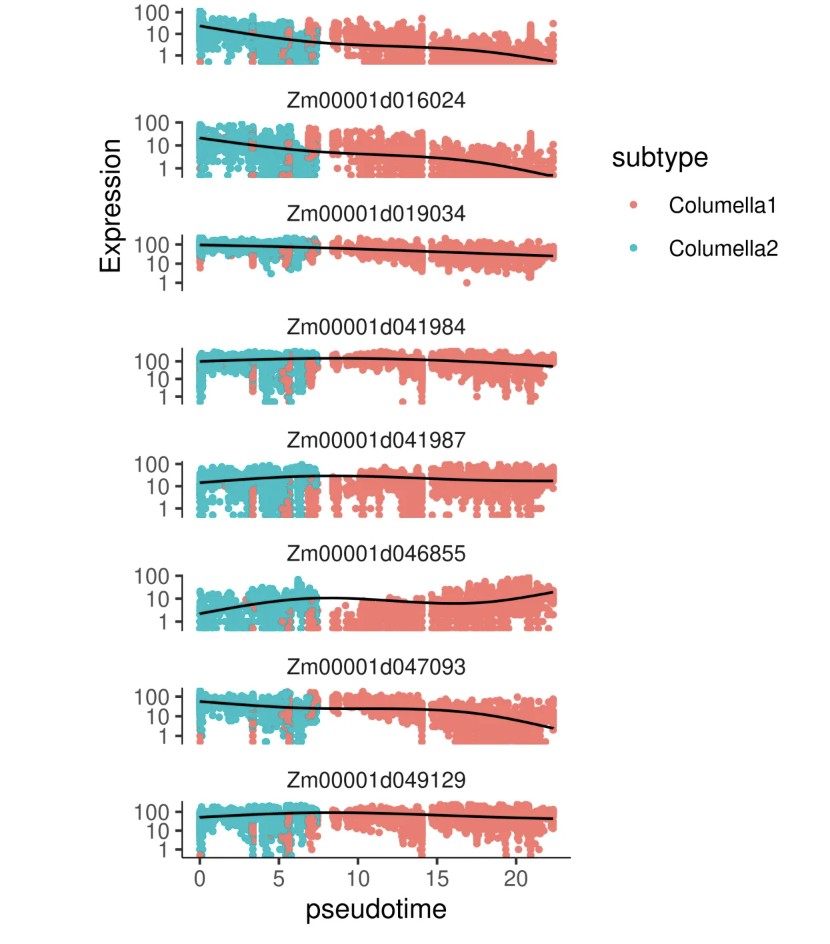
# Moran's I 检验原理:评估基因表达在轨迹上的空间自相关性 # - I > 0: 正相关,表达沿轨迹呈连续性变化 # - I < 0: 负相关,表达呈离散分布 # - I 越接近 1,基因表达随拟时序变化的趋势越强 # graph_test 识别随轨迹变化的基因 marker_test_res <- graph_test(cds, neighbor_graph = "principal_graph", cores = 20 ) # 筛选显著差异基因:q_value < 0.01 且 morans_I > 0.2 deg_sig <- marker_test_res %>% filter(q_value < 0.01 & morans_I > 0.2) %>% arrange(-morans_I) # 按 Moran's I 选择 TOP12 基因进行可视化 top12_deg <- deg_sig %>% top_n(n = 12, morans_I) %>% pull(gene_short_name) %>% as.character() # 绘制 TOP12 基因在 UMAP 上的表达分布(每个基因一个小图) plot_cells(cds, genes = top12_deg, show_trajectory_graph = FALSE, label_cell_groups = FALSE, label_leaves = FALSE ) # 绘制 TOP12 基因在拟时序中的表达趋势(折线图) plot_genes_in_pseudotime(cds[top12_deg, ], color_cells_by = "subtype", min_expr = 0.5 )
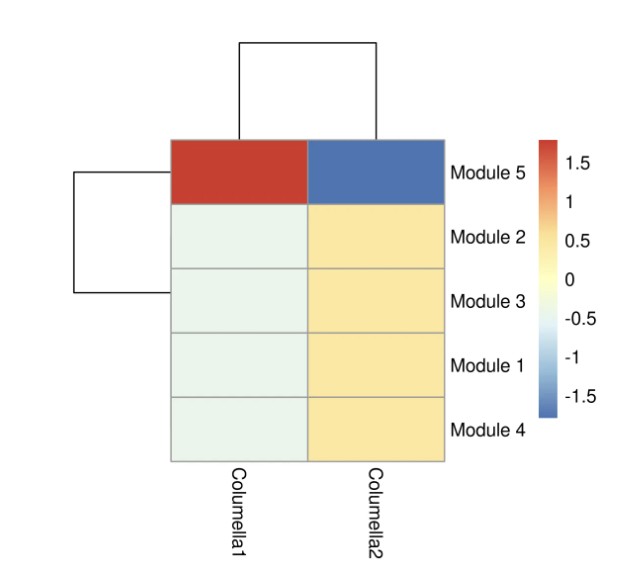
7. 基因共表达模块分析
# 获取所有显著差异基因列表
genelist <- pull(deg_sig, gene_short_name) %>% as.character()
# 构建基因共表达模块(使用 Leiden 算法聚类)
# resolution = 1e-2 控制模块粒度,值越小模块越少
gene_module <- find_gene_modules(cds[genelist, ],
resolution = 1e-2,
cores = 20
)
# 按细胞类型(subtype)聚合模块的平均表达量
cell_group <- tibble::tibble(
cell = row.names(colData(cds)),
cell_group = colData(cds)[, "subtype"]
)
agg_mat <- aggregate_gene_expression(cds, gene_module, cell_group)
row.names(agg_mat) <- stringr::str_c("Module ", row.names(agg_mat))
# 绘制模块-细胞类型表达热图(行:模块,列:细胞类型)
pheatmap(agg_mat,
scale = "column", # 按列标准化
clustering_method = "ward.D2",
main = "Gene Module Expression by Cell Type"
)
# 将模块信息添加到差异基因表
deg_sig <- cbind(deg_sig, gene_module)
8. 基因表达趋势聚类
# 提取显著差异基因的拟时序表达矩阵
genes_sig <- row.names(deg_sig)
pre_pseudotime_matrix <- getFromNamespace("pre_pseudotime_matrix", "ClusterGVis")
mat <- pre_pseudotime_matrix(cds_obj = cds, gene_list = genes_sig)
# K-means 聚类(分为 5 个趋势簇)
ck <- clusterData(obj = as.data.frame(mat),
clusterMethod = "kmeans",
clusterNum = 5
)
# 随机采样 30 个基因作为热图标记基因
markGenes <- sample(rownames(mat), 30, replace = FALSE)
9. 功能富集分析(KEGG 通路 + GO 术语)
# 初始化富集结果容器
enrichKEGG <- NULL
enrichGO <- NULL
topn <- 5
# ==================== KEGG 通路富集 ====================
# 读取 KEGG 基因-通路映射文件(玉米参考基因组注释)
gene2pathway <- read.table(
"/share/ref/Zea_mays/annotations/kegg.gene2pathway.tsv",
header = TRUE, check.names = FALSE, sep = "\t",
comment.char = "", quote = ""
)
# 构建 TERM2GENE(通路-基因映射)和 TERM2NAME(通路ID-名称映射)
TERM2GENE.KEGG <- gene2pathway[, c(2, 1)] %>% distinct()
TERM2NAME.KEGG <- gene2pathway[, c(2, 3)] %>% distinct()
# ==================== GO 生物学过程富集 ====================
# 读取 GO 基因-功能映射文件
gene2go <- read.table(
"/share/ref/Zea_mays/annotations/go.go_anno_result.tsv",
sep = "\t", header = TRUE, check.names = FALSE,
comment.char = "", quote = ""
)
# 筛选生物学过程(BP = biological_process)
gene2go <- subset(gene2go, CLASS == "biological_process")
TERM2GENE.GO <- gene2go[, 2:1] %>% distinct()
TERM2NAME.GO <- gene2go[, c(2, 3)] %>% distinct()
# ==================== 执行富集分析 ====================
# KEGG 通路富集
message("enrichCluster KEGG ownSet")
enrichKEGG <- enrichCluster(object = ck,
OrgDb = NULL,
type = "ownSet", # 使用自定义背景集
pvalueCutoff = 1, # 不设 P 值阈值
idTrans = FALSE,
TERM2GENE = TERM2GENE.KEGG,
TERM2NAME = TERM2NAME.KEGG,
topn = 200,
seed = 1314 # 固定随机种子,保证可重复性
)
# 保留每个聚类簇的前 topn 个富集通路
enrichKEGG <- enrichKEGG %>% group_by(group) %>% slice_head(n = topn)
# GO 生物学过程富集
message("enrichCluster GO ownSet")
enrichGO <- enrichCluster(object = ck,
OrgDb = NULL,
type = "ownSet",
pvalueCutoff = 1,
idTrans = FALSE,
TERM2GENE = TERM2GENE.GO,
TERM2NAME = TERM2NAME.GO,
topn = 200,
seed = 1314
)
enrichGO <- enrichGO %>% group_by(group) %>% slice_head(n = topn)
# ==================== 可视化:表达趋势 + 富集注释热图 ====================
# 预定义颜色向量(用于热图侧边标注)
gocol <- c("#C6307C", "#D0AFC4", "#4991C1", "#FE7F0E", "#89558D",
"#AFC2D9", "#435B95", "#79B99D", "#D1D097", "#C4B6D0",
"#B4C8E1", "#835B53"
)
keggcol <- c("#378F67", "#F3A5A3", "#BDA09A", "#A758E5", "#D584BE",
"#BF3D3D", "#EFBED3", "#2DABB9", "#EFBB89", "#A0D494"
)
# 根据聚类簇数量动态分配颜色
gocol <- gocol[as.integer(as.factor(enrichGO$group))]
keggcol <- keggcol[as.integer(as.factor(enrichKEGG$group))]
# 绘制组合热图(左:表达趋势聚类,右:GO/KEGG 富集注释)
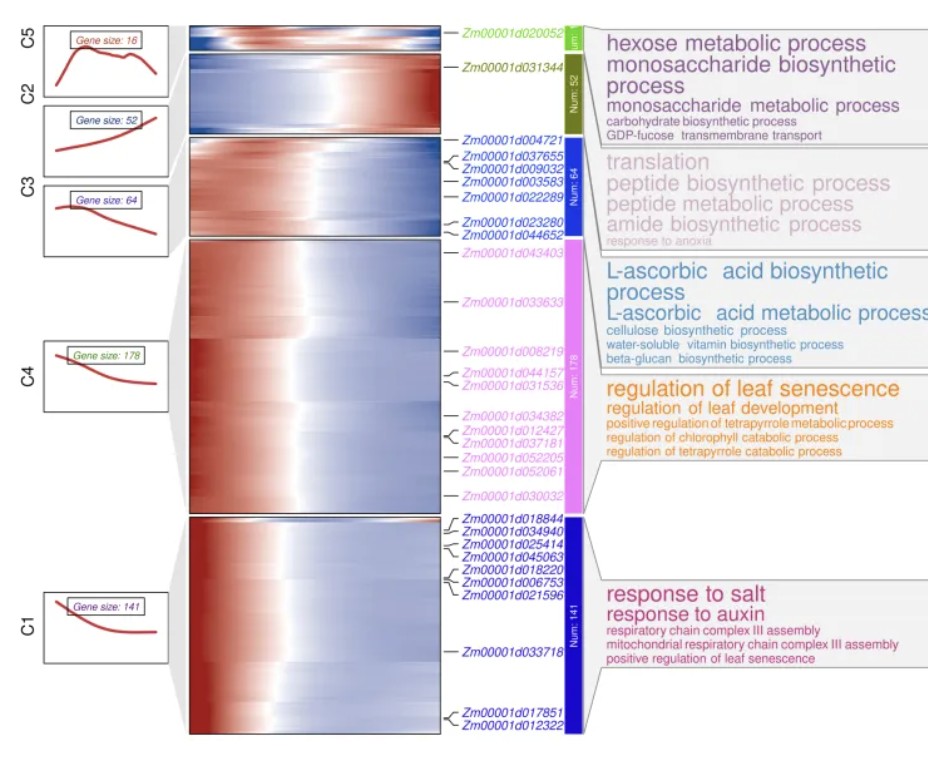
visCluster(object = ck,
plotType = "both", # 同时显示表达热图和富集注释
annoTermData = enrichGO, # GO 富集注释
annoKeggData = enrichKEGG, # KEGG 富集注释
htColList = list(
col_range = c(-2, 0, 2), # 热图颜色范围
col_color = c("#08519C", "white", "#A50F15")
),
lineSide = "left", # 富集标注放在左侧
show_row_dend = FALSE, # 不显示行树状图
addSampleAnno = FALSE, # 不添加样本注释
goSize = "pval", # 气泡大小映射 P 值
keggSize = "pval",
goCol = gocol, # GO 标注颜色
keggCol = keggcol, # KEGG 标注颜色
markGenes = markGenes # 标记基因列表
)
下次我们介绍如何手动选取轨迹进行分析。
如果遇到软件安装错误无法运行,可以使用组学大讲堂云服务器,已经安装好所有单细胞转录组数据分析的软件运行环境,省去环境配置麻烦,详情可扫下方二维码咨询:

Wang, T., Wang, F., Deng, S. et al. Single-cell transcriptomes reveal spatiotemporal heat stress response in maize roots. Nat Commun16, 177 (2025). https://doi.org/10.1038/s41467-024-55485-3
- 发表于 2026-04-13 10:19
- 阅读 ( 749 )
- 分类:转录组
