单细胞拟时序分析monocle2-代码
接之前分析:1.玉米根热胁迫单细胞转录组复现2.玉米根单细胞转录组细胞类型注释代码3.热胁迫处理玉米根单细胞转录组差异基因分析
今天来分析一下文章中用monocle2分析细胞发育部分:作者通过Monocle2拟时序分析发现,玉米根中的柱细胞可分为两个转录组特征不同的亚型(Columella 1 和 Columella 2),其中Columella 1主要分布在发育轨迹的分支2及前分支阶段,而Columella 2则集中在分支1。进一步的分支点表达分析鉴定出1997个差异表达基因,分为三类:分支2高表达的基因富集于应激响应功能,分支1高表达的基因与蛋白质代谢相关,前分支高表达的基因则参与葡萄糖代谢。尽管柱细胞已知参与低温胁迫响应,但热激处理并未改变这两个亚型的发育轨迹。总之,拟时序分析揭示了柱细胞发育的动态过程及细胞状态转换中关键基因的表达重编程。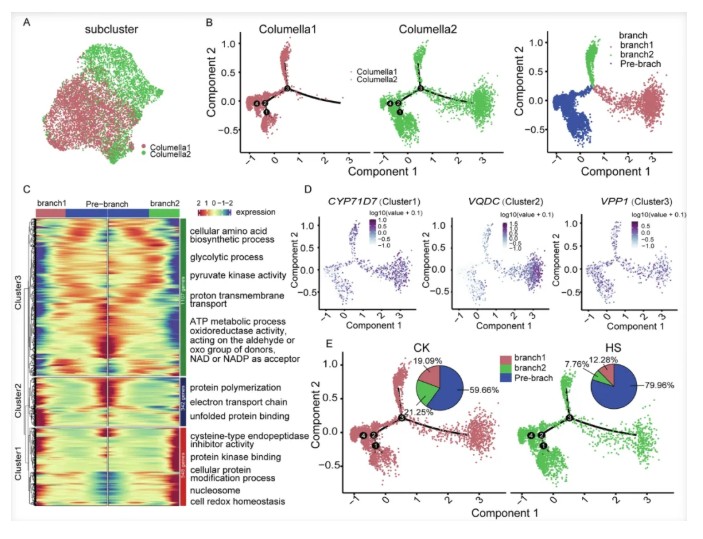
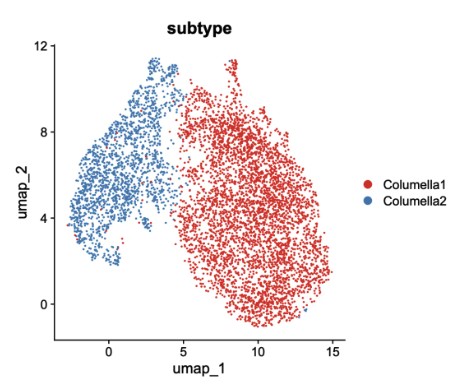
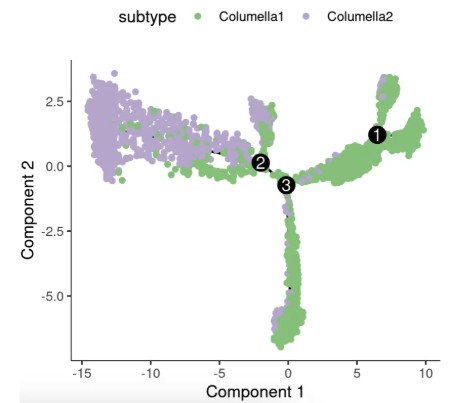
接下来我们开始分析,下面是我们分析的:Columella细胞的umap图,也是分成了两大类:
下面开始代码分析:加载需要的包
library(Seurat)
library(monocle)
library(ggpubr)
library(RColorBrewer)
library(ggplot2)
library(dplyr)
library("ClusterGVis")
library(viridis)
library(qs)
qual_col_pals <- brewer.pal.info[brewer.pal.info$category == 'qual', ]
# 处理后有73种差异还比较明显的颜色,基本够用
col_vector <- unlist(mapply(brewer.pal,
qual_col_pals$maxcolors,
rownames(qual_col_pals)))
1. 数据加载与预处理
# 使用qread函数快速加载已保存的Seurat对象(.qs格式),支持多线程
# 该对象应该是已经过注释的柱状细胞(Columella)亚型数据
scRNA <- qread("../06.subset/Columella.added.subtype.qs", nthreads = 4)
# 提取原始RNA测序的counts矩阵(非标准化数据)
# LayerData是较新Seurat版本中的推荐方法,替代直接访问@assays槽
# layer = "counts" 确保使用UMI/原始计数,这是Monocle2要求的输入类型
expr_matrix <- LayerData(
object = scRNA,
assay = "RNA",
layer = "counts"
)
# 旧版代码(已注释):直接从Seurat对象中提取counts矩阵
# expr_matrix <- as(as.matrix(scRNA@assays[[assay]]@counts), 'sparseMatrix')
# 提取细胞的元数据(表型信息),例如细胞类型、样本来源、批次等
# 这些信息将用于后续的可视化和条件矫正
p_data <- scRNA@meta.data
head(p_data)
# 准备基因特征数据(f_data)
# Monocle2要求f_data至少包含一列 'gene_short_name',用于标记基因
# 这里使用基因的row.names作为基因短名
f_data <- data.frame(
gene_short_name = row.names(scRNA),
row.names = row.names(scRNA)
)
# 数据维度检查(注释):
# expr_matrix 的行数 == f_data 的行数(基因数)
# expr_matrix 的列数 == p_data 的行数(细胞数)
2. 构建Monocle2的CDS对象
# 将普通data.frame转换为Bioconductor的AnnotatedDataFrame对象
# 这是Monocle2对象结构的要求
pd <- new('AnnotatedDataFrame', data = p_data) # 表型数据
fd <- new('AnnotatedDataFrame', data = f_data) # 特征(基因)数据
# 核心步骤:创建CellDataSet对象
# lowerDetectionLimit: 低于0.5的表达式量被视为未检测到
# expressionFamily: 指定数据分布类型。negbinomial.size() 适用于UMI计数数据(负二项分布)
cds <- newCellDataSet(
expr_matrix,
phenoData = pd,
featureData = fd,
lowerDetectionLimit = 0.5,
expressionFamily = negbinomial.size()
)
# 估计每个细胞的文库大小(size factors),用于标准化
cds <- estimateSizeFactors(cds)
# 估计基因的离散度(variance-mean关系),对于负二项分布模型至关重要
cds <- estimateDispersions(cds)
3. 选择用于构建轨迹的基因
# 释放原始Seurat对象的内存,因为后续只使用CDS对象
rm(scRNA)
# 获取离散度表格:包含每个基因的平均表达量、拟合离散度、经验离散度
disp_table <- dispersionTable(cds)
# 筛选高变基因(dispersion.genes)作为排序基因
# 条件1: mean_expression >= 0.1 (基因平均表达量不能太低)
# 条件2: dispersion_empirical >= 1 * dispersion_fit (经验离散度不低于拟合值,即高变)
# 这些基因将驱动后续的细胞排序(轨迹推断)
disp.genes <- subset(
disp_table,
mean_expression >= 0.1 &
dispersion_empirical >= 1 * dispersion_fit
)$gene_id
# 将筛选出的高变基因设置为排序基因(ordering genes)
cds <- setOrderingFilter(cds, disp.genes)
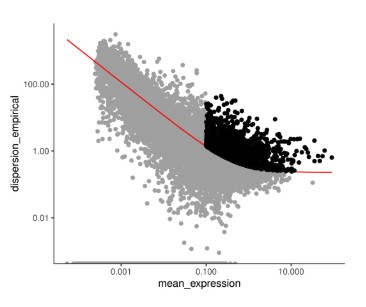
# 可视化排序基因:展示经验离散度 vs 平均表达量,高亮选中的基因
plot_ordering_genes(cds)
4. 降维(Dimensionality Reduction)
# 关键降维函数 reduceDimension
# max_components = 2: 最终降维到2维空间(便于可视化)
# residualModelFormulaStr = "~SampleID": 剔除样本ID(SampleID)的批次效应
# reduction_method = "DDRTree": 使用Monocle2特有的反向图嵌入树算法
# num_dim = 50: 在DDRTree内部计算时保留的隐含维度(通常2-100之间,越大信息保留越多但计算越慢)
cds <- reduceDimension(
cds,
max_components = 2,
residualModelFormulaStr = "~SampleID",
reduction_method = "DDRTree",
num_dim = 50
)
5. 拟时间排序
message("orderCells")
# orderCells: 核心排序函数,基于DDRTree的降维结果构建最小生成树,并计算每个细胞的拟时间
# 注意:orderCells会自动选择轨迹的一个端点作为“起点”(拟时间为0)
cds <- orderCells(cds)
6. 可视化结果
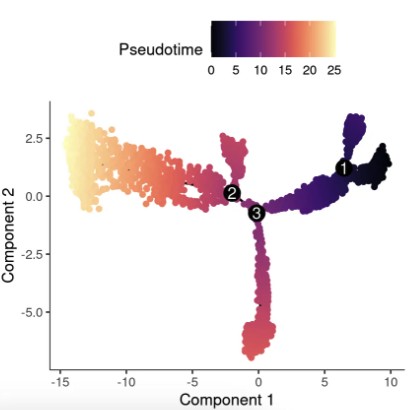
6.1 拟时间(Pseudotime)
# 标准轨迹图:颜色表示拟时间(通常从紫色到黄色代表时间进程)
# 注意:plot_cell_trajectory 默认使用DDRTree降维坐标(reducedDimS)
plot_cell_trajectory(cds, color_by = "Pseudotime") +
scale_color_viridis(option = "magma") # magma色系,拟时间常用
6.2 细胞状态(State)可视化
# State是Monocle2基于轨迹分支识别出的不同“命运”或亚群(通常为整数编号)
plot_cell_trajectory(cds, color_by = "State") + scale_color_manual(values = col_vector) # col_vector需提前定义的自定义颜色向量
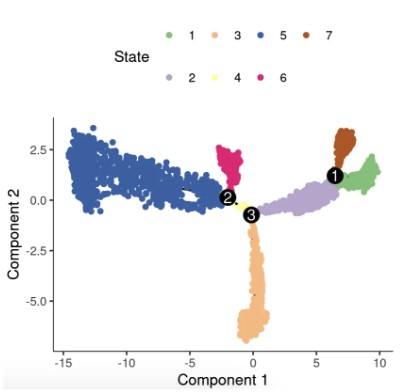
6.3 细胞亚群(subtype)可视化
检查已有的注释亚群在轨迹上的分布,观察起始细胞是否符合预期
plot_cell_trajectory(cds, color_by = "subtype") + scale_colour_manual(values = col_vector)
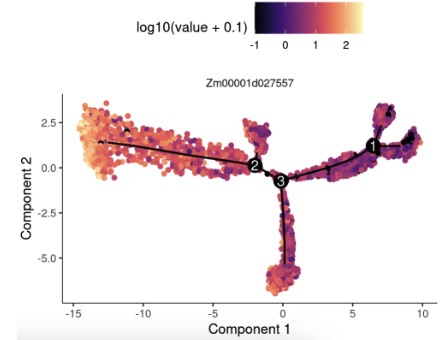
6.4 基因降维图可视化
检查关键基因表达变化
plot_cell_trajectory(cds, markers = "Zm00001d027557",use_color_gradient=T)+ scale_color_viridis(option="magma")
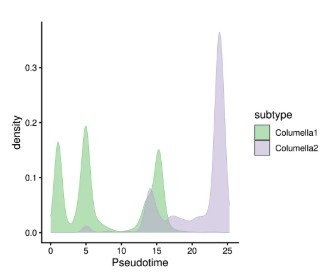
6.5 拟时间密度分布(拟时间 vs 细胞密度)
查看不同细胞亚群在拟时间轴上的富集区域
# 提取拟时间排序信息表
df <- data.frame(
barcode = row.names(pData(cds)),
pData(cds)
)
# 绘制密度图:x轴为Pseudotime,y轴为密度,不同颜色代表不同subtype
ggplot(df, aes(Pseudotime, colour = subtype, fill = subtype)) +
geom_density(bw = 0.5, size = 0.2, alpha = 0.5) +
theme_classic2() + # 使用经典主题(可能需要加载ggpubr或自定义)
scale_colour_manual(values = col_vector) +
scale_fill_manual(values = col_vector) +
labs(fill = "subtype") +
guides(color = "none") # 隐藏颜色图例(因为fill已足够)
分支分析,这里我们分析branch3:
# ==================== BEAM: 分支表达分析 ====================
# BEAM (Branch Expression Analysis Modeling) 用于识别在细胞命运决定点(分支点)处
# 表现出显著差异表达的基因,这些基因可能驱动细胞向不同方向分化
#
# 参数说明:
# - cds[disp.genes,]: 使用之前筛选出的高变基因(disp.genes)进行BEAM分析
# - branch_point: 指定要分析的分支点编号(例如1、2等,取决于轨迹结构)
# - branch_states: 指定要比较的两个分支状态(例如c(1,2)表示比较State1和State2)
# - cores: 并行计算使用的CPU核心数
BEAM_res <- BEAM(
cds[disp.genes, ],
branch_point = 3,
branch_states = NULL,
cores = 5
)
# 注意:此步骤运行较慢,因为BEAM需要对每个基因拟合统计模型
# 实际运行时间取决于基因数量、细胞数量和分支复杂度
# ==================== 排序和筛选BEAM结果 ====================
# 按q值(校正后的p值,FDR)升序排序,最显著的基因排在前面
BEAM_res <- BEAM_res[order(BEAM_res$qval), ]
# 只保留三列关键信息:基因名称、p值、校正后的q值
BEAM_res <- BEAM_res[, c("gene_short_name", "pval", "qval")]
# 保存BEAM分析结果到文件
# 输出路径和文件名由opt列表中的outdir和prefix参数决定
write.table(
BEAM_res,
file = "BEAM.DEGenes.tsv",
sep = "\t",
quote = FALSE,
row.names = FALSE,
col.names = TRUE
)
# ==================== 筛选用于热图展示的显著基因 ====================
# 根据设定的q值阈值(例如0.05或0.01)筛选显著差异表达基因
BEAM_res_sig <- subset(BEAM_res, qval < 0.001)
# 从CDS对象中提取显著基因的表达数据(用于后续热图绘制)
cds_subset <- cds[row.names(BEAM_res_sig), ]
# ==================== 绘制分支表达热图 ====================
# plot_genes_branched_heatmap2: Monocle2中专门用于分支分析的增强热图函数
# 该函数会:
# 1. 提取指定分支点处的基因表达数据
# 2. 将细胞按拟时间和分支状态排序
# 3. 对基因进行聚类
# 4. 生成热图展示基因在两个分支中的表达模式
df <- plot_genes_branched_heatmap2(
cds_subset,
# 指定要分析的分支点
branch_point = opt$branch_point,
# 热图中基因聚类的数量(例如4-6个簇)
num_clusters = 4,
# 分支标签(用于图例,例如c("Branch1", "Branch2"))
branch_labels = c("Cell fate 1", "Cell fate 2"),
# 要比较的两个状态(必须与branch_point互斥)
# 这里使用branch_states而非branch_point
branch_states = NULL,
# 分支的颜色设置
branch_colors = c("#979797", "#F05662", "#7990C8"),
# 并行计算核心数
cores = 4,
# 是否使用基因短名称(TRUE则显示常见基因名)
use_gene_short_name = TRUE,
# 是否在热图中显示行名(基因名)
show_rownames = TRUE,
# 是否返回热图对象(FALSE表示直接绘制)
return_heatmap = FALSE
)
# df 返回值通常包含以下组件:
# - wide.res: 宽格式的基因表达矩阵(基因 × 细胞)
# - gene: 基因列表
# - cluster: 基因聚类分配结果
# - cell: 细胞排序信息
非模式动植物拟时间热图,并做GOKEGG
# ==================== 1. 初始化KEGG和GO的数据容器 ====================
# 这些变量将在后续存储通路信息
TERM2GENE.KEGG = NULL # KEGG: 通路与基因的对应关系(两列:通路ID, 基因ID)
TERM2NAME.KEGG = NULL # KEGG: 通路ID与通路名称的对应关系
TERM2GENE.GO = NULL # GO: 通路与基因的对应关系
TERM2NAME.GO = NULL # GO: 通路ID与通路名称的对应关系
# ==================== 2. 加载并处理KEGG通路数据 ====================
# 读取KEGG基因-通路映射文件
# 文件格式预期:第1列是基因ID,第2列是通路ID,第3列是通路名称
gene2pathway <- read.table(
"kegg.gene2pathway.tsv",
header = TRUE, # 第一行为列名
check.names = FALSE, # 不自动检查列名
sep = "\t", # 制表符分隔
comment.char = "", # 不忽略注释行(全部读取)
quote = "" # 不处理引号
)
# 构建通路-基因映射表(TERM2GENE)
# 选择第2列(通路ID)和第1列(基因ID),并去重
TERM2GENE.KEGG <- gene2pathway[, c(2, 1)] %>% distinct()
# 构建通路ID-通路名称映射表(TERM2NAME)
# 选择第2列(通路ID)和第3列(通路名称),并去重
TERM2NAME.KEGG <- gene2pathway[, c(2, 3)] %>% distinct()
# ==================== 3. 加载并处理GO通路数据 ====================
# 读取GO注释结果文件(通常来自clusterProfiler或类似工具)
# 文件格式预期:包含基因ID、GO ID、GO名称、分类类型(CLASS)等
gene2go <- read.table(
"go.go_anno_result.tsv",
sep = "\t",
header = TRUE,
check.names = FALSE,
comment.char = "",
quote = ""
)
# 指定要分析的GO类别(可选项:BP、CC、MF)
go_type <- "biological_process" # BP: 生物学过程
# 筛选特定类别的GO注释(这里选择BP)
message(sprintf("subset gene2go by go_type %s", "BP"))
gene2go <- subset(gene2go, CLASS == "BP")
# 构建GO通路-基因映射表
# 第2列(GO ID)和第1列(基因ID)
TERM2GENE.GO <- gene2go[, 2:1] %>% distinct()
# 构建GO ID-通路名称映射表
# 第2列(GO ID)和第3列(通路名称)
TERM2NAME.GO <- gene2go[, c(2, 3)] %>% distinct()
# ==================== 4. 执行KEGG通路富集分析 ====================
message("enrichCluster KEGG ownSet")
# enrichCluster函数:对每个基因模块(cluster)分别进行通路富集
# 注意:这里的df对象应该是包含基因模块信息的数据结构(如hclust结果)
enrichKEGG <- enrichCluster(
object = df,
OrgDb = NULL, # 不需要内置OrgDb(使用自定义通路集)
type = "ownSet", # 使用用户自定义的通路集(而非KEGG在线数据库)
pvalueCutoff = 1, # 不筛选p值(保留所有结果)
idTrans = FALSE, # 不需要ID转换(已经是标准ID)
TERM2GENE = TERM2GENE.KEGG, # 自定义的通路-基因映射
TERM2NAME = TERM2NAME.KEGG, # 自定义的通路ID-名称映射
topn = 200, # 每个模块保留前200个通路
seed = 1314 # 设置随机种子(确保可重复性)
)
# 对每个基因模块(group),只保留前topn个显著通路(按p值排序)
enrichKEGG <- enrichKEGG %>% group_by(group) %>% slice_head(n = topn)
# ==================== 5. 执行GO通路富集分析 ====================
# 逻辑与KEGG类似,但使用GO数据集
enrichGO <- enrichCluster(
object = df,
OrgDb = NULL,
type = "ownSet",
pvalueCutoff = 1,
idTrans = FALSE,
TERM2GENE = TERM2GENE.GO, # 使用GO映射数据
TERM2NAME = TERM2NAME.GO,
topn = 200,
seed = 1314
)
# 同样只保留每个组的前200个通路
enrichGO <- enrichGO %>% group_by(group) %>% slice_head(n = topn)
# ==================== 6. 为不同模块分配可视化颜色 ====================
# 定义GO富集分析的颜色向量(每个基因模块对应一个颜色)
gocol <- c(
"#C6307C", "#D0AFC4", "#4991C1", "#FE7F0E", "#89558D",
"#AFC2D9", "#435B95", "#79B99D", "#D1D097", "#C4B6D0",
"#B4C8E1", "#835B53"
)
# 定义KEGG富集分析的颜色向量
keggcol <- c(
"#378F67", "#F3A5A3", "#BDA09A", "#A758E5", "#D584BE",
"#BF3D3D", "#EFBED3", "#2DABB9", "#EFBB89", "#A0D494"
)
# 根据enrichGO中实际出现的模块数量,重新索引颜色
# as.integer(as.factor()) 将模块名称转换为整数索引
gocol <- gocol[as.integer(as.factor(enrichGO$group))]
keggcol <- keggcol[as.integer(as.factor(enrichKEGG$group))]
# ==================== 7. 随机选择展示的基因 ====================
# 从df$wide.res$gene中随机抽取30个基因用于热图标记
# wide.res通常包含基因表达矩阵或模块分配信息
gene <- sample(df$wide.res$gene, 30, replace = FALSE)
# ==================== 8. 可视化富集分析结果 ====================
# visCluster: 综合可视化函数,通常显示:
# - 左侧:基因模块的聚类热图
# - 右侧:GO和KEGG通路的富集结果(气泡图或条形图)
visCluster(
object = df,
plotType = "both", # 同时显示热图和富集分析结果
# 热图颜色设置(三色渐变)
htColList = list(
col_range = c(-2, 0, 2), # 数值范围
col_color = c("#08519C", "white", "#A50F15") # 蓝-白-红
),
# GO富集结果数据
annoTermData = enrichGO,
# KEGG富集结果数据
annoKeggData = enrichKEGG,
lineSide = "left", # 模块标签显示在左侧
goSize = "pval", # GO气泡图大小映射到p值(常用-log10(p))
keggSize = "pval", # KEGG气泡图大小映射到p值
# 颜色向量
goCol = gocol, # GO各模块的颜色
keggCol = keggcol, # KEGG各模块的颜色
show_row_dend = FALSE, # 不显示行聚类树(避免图形过于复杂)
markGenes = gene # 在热图中标记这30个基因
)
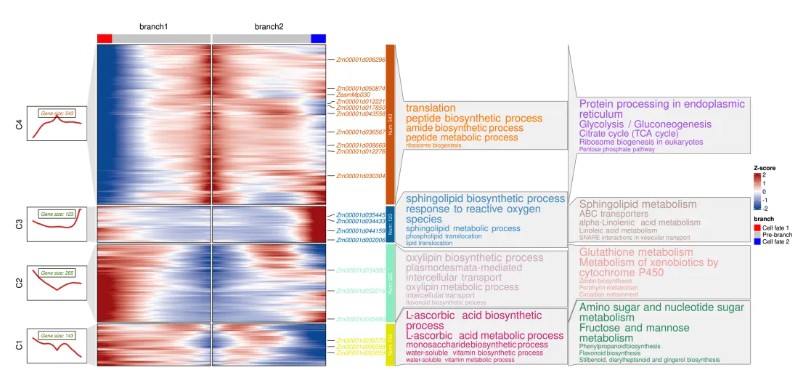
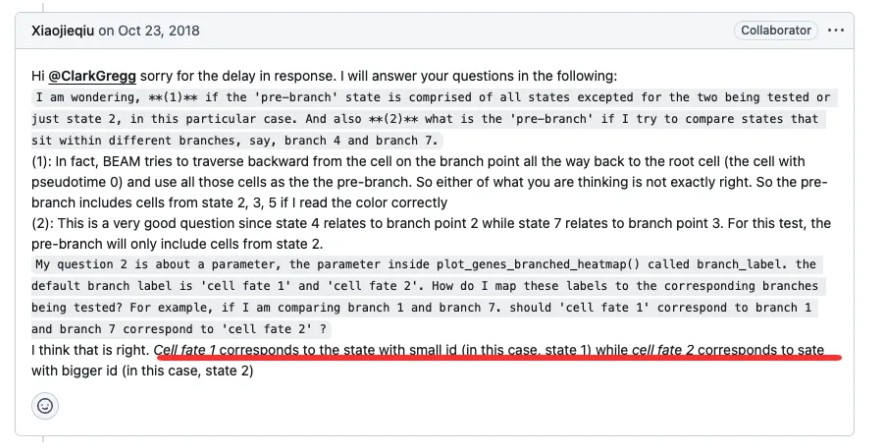
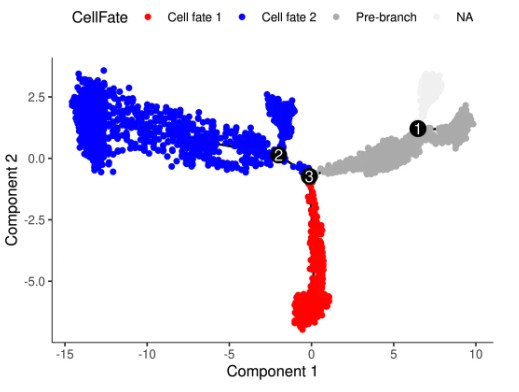
monocle2中分析不同branch,不同的cell fate 指代哪部分细胞呢?很多同学遇到这个问题,我这边也是很遗憾。这里也有作者给出的信息https://github.com/cole-trapnell-lab/monocle-release/issues/219,总结起来就是沿着拟时间到branch点之前的为prebranch,较小的state为cell fate1 较大的state为cell fate2:
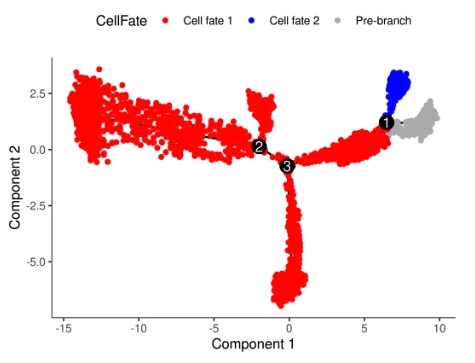
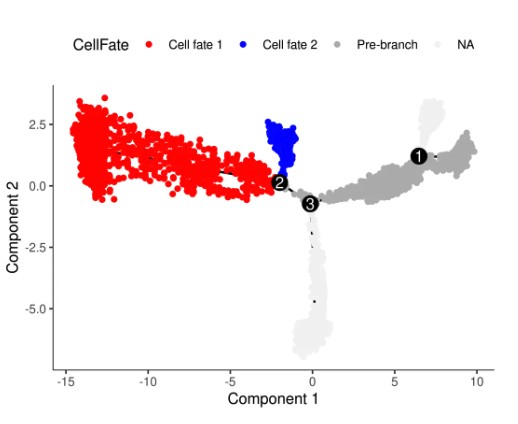
但是,根据上面的信息,如果自己的图中的 branch point 特别多,就不是很好区分了,为了方便大家区分不同branch细胞来源,这里分别用不同颜色标记出来 方便大家理解,NA表示不涉及的细胞。
如果分析:branch1
如果分析:branch2
如果分析:branch3
如果遇到软件安装错误无法运行,可以使用组学大讲堂云服务器,已经安装好所有单细胞转录组数据分析的软件运行环境,省去环境配置麻烦,详情可扫下方二维码咨询:

Wang, T., Wang, F., Deng, S. et al. Single-cell transcriptomes reveal spatiotemporal heat stress response in maize roots. Nat Commun16, 177 (2025). https://doi.org/10.1038/s41467-024-55485-3
- 发表于 2026-04-10 16:36
- 阅读 ( 1460 )
- 分类:转录组
