玉米根单细胞转录组细胞类型注释代码
玉米根单细胞转录组细胞类型注释代码
接上回细胞聚类分群结果:玉米根热胁迫单细胞转录组复现这次我们将聚类结果进行单细胞注释,首先收集细胞类型特异的marker基因,也可以通过植物细胞类型marker基因数据库查找,例如:PCMDB 数据库(https://www.tobaccodb.org/pcmdb/homePage)包含 6 个植物物种(拟南芥、玉米、水稻、大豆、番茄、烟草)的 Marker 基因,涉及 263 种细胞类型。其中3,119 个标记基因通过实验验证可靠。PsctH数据库(http://jinlab.hzau.edu.cn/PsctH/)收集了 5 个物种(拟南芥、玉米、水稻、花生、番茄)Marker 基因,涉及 26 种细胞类型。PlantscRNAdb数据库(http://ibi.zju.edu.cn/plantscrnadb/)包括了33 个物种(拟南芥、水稻、番茄、玉米、草莓、杨树、烟草、浮萍、白菜、木薯、蒺藜苜蓿、裂叶荆芥、陆地棉、大豆、蝴蝶兰等)的 Marker 基因。因此,模式植物细胞类型的标记基因相对完善。但是针对大多数非模式植物物种,可用的细胞类型标记基因非常少,无法确定这些物种细胞类型将难以进行后续分析。但是可以利用生物信息学方法,跨物种寻找同源基因来定位标记基因。下面是我收集的玉米根不同细胞类型标记基因:
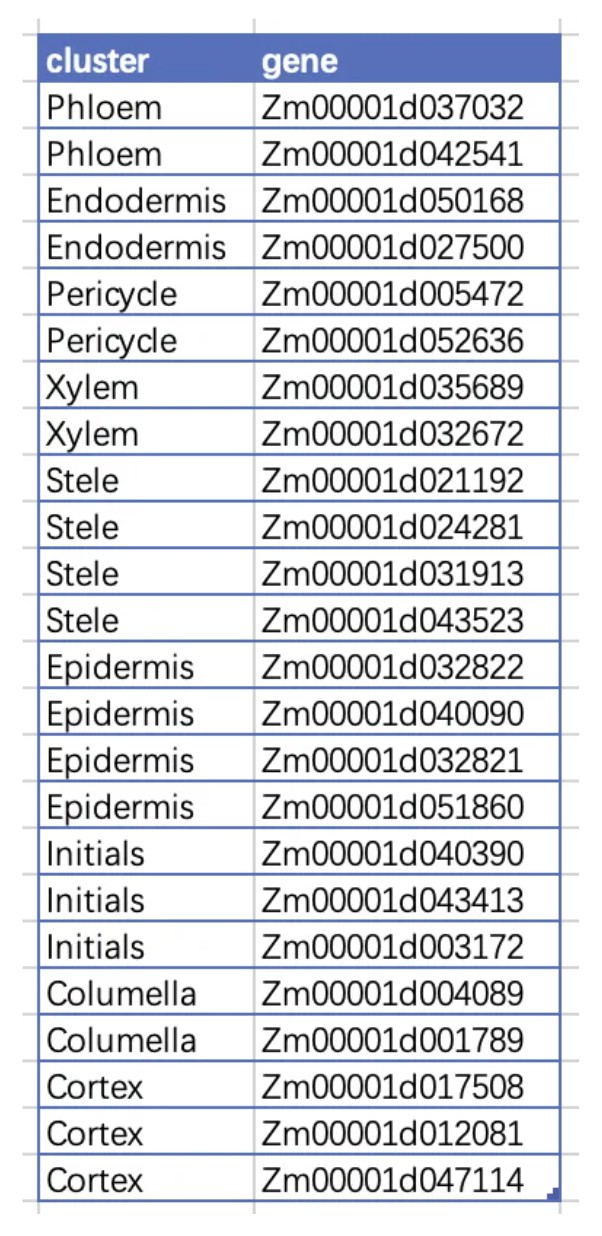
玉米根细胞类型注释MARKER
收集玉米细胞亚型标记基因:
| 细胞类型 | 中文翻译 | 基因名称及编号 |
| Phloem | 韧皮部 | Zm00001d037032, Zm00001d042541 |
| Endodermis | 内皮层 | LCR74 (Zm00001d050168), Zm00001d027500 |
| Pericycle | 周鞘 | Zm00001d005472, Zm00001d052636 |
| Xylem | 木质部 | MYB46 (Zm00001d038878), Zm00001d035689, Zm00001d032672 |
| Stele | 中柱(维管柱) | WOL1 (Zm00001d014297), TMO5 (Zm00001d004630), Zm00001d021192, Zm00001d024281, Zm00001d031913, Zm00001d043523 |
| Epidermis | 表皮 | RHD6 (Zm00001d017804), Zm00001d032822, Zm00001d040090, Zm00001d032821, Zm00001d051860 |
| Initials | 初始细胞(分生组织起始细胞) | Zm00001d040390, Zm00001d043413, Zm00001d003172 |
| Columella | 柱状细胞(根冠柱状细胞) | Zm00001d004089, Zm00001d001789 |
| Cortex | 皮层 | CO2 (Zm00001d051384), CO2-like (Zm00001d027291), Zm00001d017508, Zm00001d012081, Zm00001d047114 |
整理成表格方便R读入(marker.tsv):
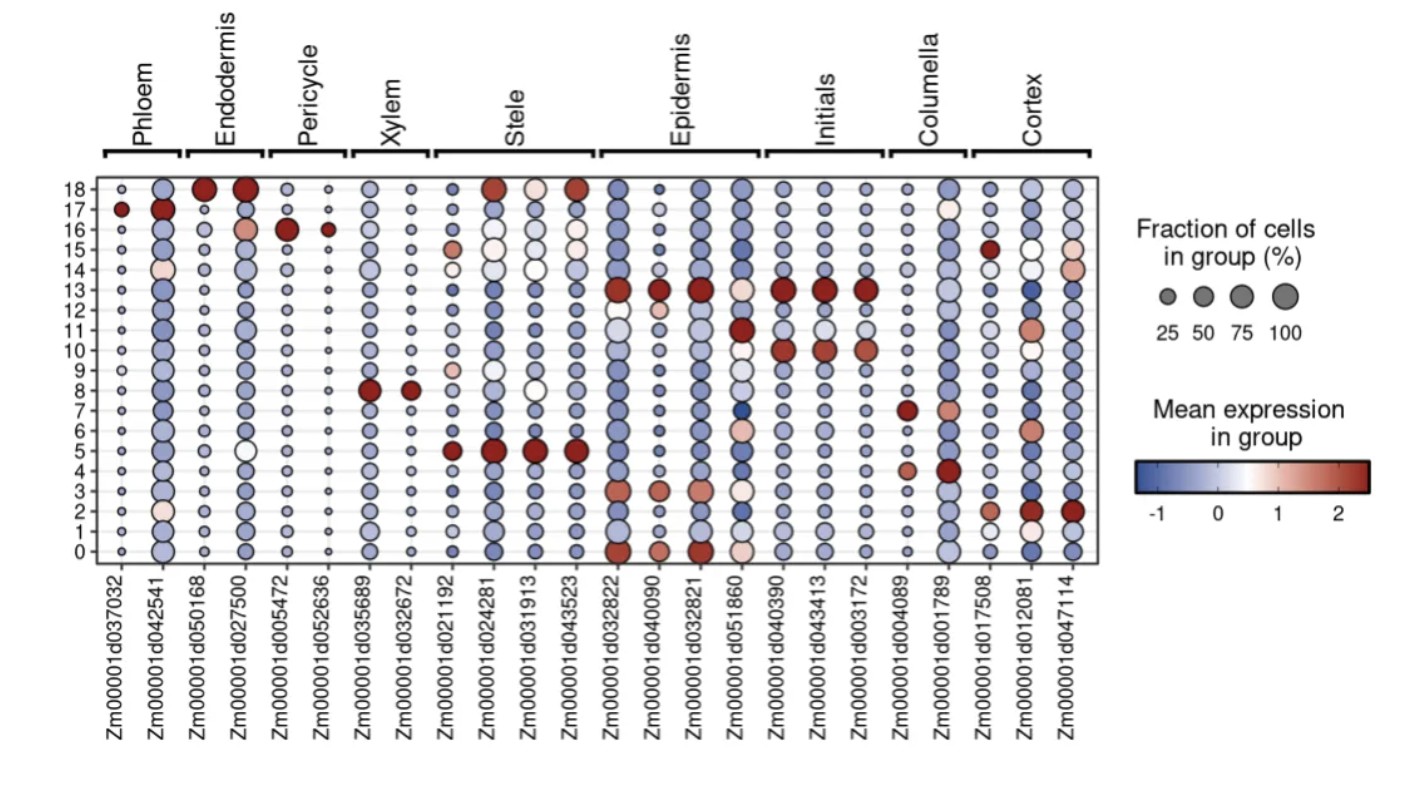
展示maker基因在不同cluster亚群表达情况,运行下面的代码出图:
library(Seurat) library("tidyverse") library(qs) library(scop) library(scRNAtoolVis) # 读入前面聚类结果seurat对象qs文件 obj <- qread("03.seurat_cluster/root.qs", nthreads = 4) # 准备细胞和gene对应关系 marker_df <- data.frame( cluster = c( "Phloem", "Phloem", "Endodermis", "Endodermis", "Pericycle", "Pericycle", "Xylem", "Xylem", "Stele", "Stele", "Stele", "Stele", "Epidermis", "Epidermis", "Epidermis", "Epidermis", "Initials", "Initials", "Initials", "Columella", "Columella", "Cortex", "Cortex", "Cortex" ), gene = c( "Zm00001d037032", "Zm00001d042541", "Zm00001d050168", "Zm00001d027500", "Zm00001d005472", "Zm00001d052636", "Zm00001d035689", "Zm00001d032672", "Zm00001d021192", "Zm00001d024281", "Zm00001d031913", "Zm00001d043523", "Zm00001d032822", "Zm00001d040090", "Zm00001d032821", "Zm00001d051860", "Zm00001d040390", "Zm00001d043413", "Zm00001d003172", "Zm00001d004089", "Zm00001d001789", "Zm00001d017508", "Zm00001d012081", "Zm00001d047114" ) ) # 细胞类型顺序指定 marker_df$cluster <- factor( marker_df$cluster, levels = c( "Phloem", "Endodermis", "Pericycle", "Xylem", "Stele", "Epidermis", "Initials", "Columella", "Cortex" ) ) # 绘图 jjDotPlot( object = obj, markerGene = marker_df, anno = TRUE, plot.margin = c(3.5, 1, 1, 1), id = "seurat_clusters", ytree = FALSE, dot.col = c("#08519C", "white", "#A50F15") )
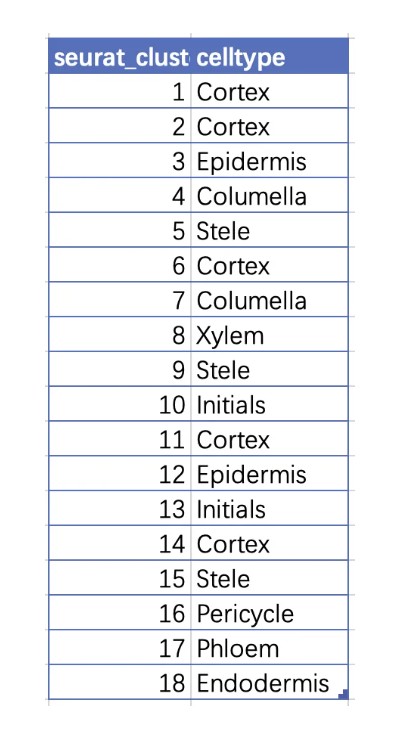
添加细胞类型注释信息到seurat对象中 ,并绘图展示结果

# 自定义颜色,满足个人审美,不同图片细胞颜色分配统一
# 这里大家可自己设置喜欢的颜色
#################################
dimCols <- c(
"#E41A1C", "#377EB8", "#4DAF4A", "#984EA3", "#FF7F00",
"#A65628", "#F781BF", "#999999", "#66C2A5", "#FC8D62",
"#8DA0CB", "#E78AC3", "#A6D854", "#FFD92F", "#E5C494",
"#B3B3B3", "#7FC97F", "#BEAED4", "#FDC086", "#386CB0",
"#F0027F", "#BF5B17", "#666666", "#1B9E77", "#D95F02",
"#7570B3", "#E7298A", "#66A61E", "#E6AB02", "#A6761D",
"#666666", "#A6CEE3", "#1F78B4", "#B2DF8A", "#33A02C",
"#FB9A99", "#E31A1C", "#FDBF6F", "#FF7F00", "#CAB2D6",
"#6A3D9A", "#FFFF99", "#B15928", "#8DD3C7", "#FFFFB3",
"#BEBADA", "#FB8072", "#80B1D3", "#FDB462", "#B3DE69",
"#FCCDE5", "#D9D9D9", "#BC80BD", "#CCEBC5", "#FFED6F",
"#FBB4AE", "#B3CDE3", "#CCEBC5", "#DECBE4", "#FED9A6",
"#FFFFCC", "#E5D8BD", "#FDDAEC", "#F2F2F2", "#B3E2CD",
"#FDCDAC", "#CBD5E8", "#F4CAE4", "#E6F5C9", "#FFF2AE",
"#F1E2CC", "#CCCCCC"
)
# cluster 命名细胞名字
new.cluster.ids <- c(
"0" = "Epidermis", "1" = "Cortex", "2" = "Cortex",
"3" = "Epidermis", "4" = "Columella", "5" = "Stele",
"6" = "Cortex", "7" = "Columella", "8" = "Xylem",
"9" = "Stele", "10" = "Initials", "11" = "Cortex",
"12" = "Epidermis", "13" = "Initials", "14" = "Cortex",
"15" = "Stele", "16" = "Pericycle", "17" = "Phloem",
"18" = "Endodermis"
)
obj <- RenameIdents(obj, new.cluster.ids)
# 添加细胞名字分组列
obj$celltype <- Idents(obj)
# Umap图
CellDimPlot(
obj,
group.by = "celltype",
reduction = "umap",
palcolor = dimCols,
theme_use = "theme_blank"
)
# tsne图展示
CellDimPlot(
obj,
group.by = "celltype",
reduction = "tsne",
palcolor = dimCols,
theme_use = "theme_blank"
)
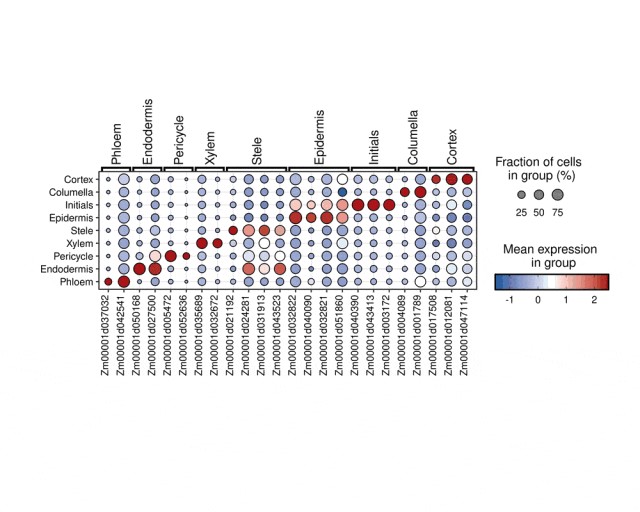
# 重新绘制 marker基因dotplot结果图,用于文章展示
celltype_df <- data.frame(
cluster = c(
"Phloem", "Phloem", "Endodermis", "Endodermis",
"Pericycle", "Pericycle", "Xylem", "Xylem",
"Stele", "Stele", "Stele", "Stele",
"Epidermis", "Epidermis", "Epidermis", "Epidermis",
"Initials", "Initials", "Initials",
"Columella", "Columella",
"Cortex", "Cortex", "Cortex"
),
gene = c(
"Zm00001d037032", "Zm00001d042541", "Zm00001d050168",
"Zm00001d027500", "Zm00001d005472", "Zm00001d052636",
"Zm00001d035689", "Zm00001d032672", "Zm00001d021192",
"Zm00001d024281", "Zm00001d031913", "Zm00001d043523",
"Zm00001d032822", "Zm00001d040090", "Zm00001d032821",
"Zm00001d051860", "Zm00001d040390", "Zm00001d043413",
"Zm00001d003172", "Zm00001d004089", "Zm00001d001789",
"Zm00001d017508", "Zm00001d012081", "Zm00001d047114"
)
)
# 细胞类型排序,这个需要自己指定一下
celltype.order <- c(
"Phloem", "Endodermis", "Pericycle", "Xylem",
"Stele", "Epidermis", "Initials", "Columella", "Cortex"
)
celltype_df$cluster <- factor(celltype_df$cluster, levels = celltype.order)
# 绘图
jjDotPlot(
object = obj,
markerGene = celltype_df,
anno = TRUE,
plot.margin = c(3, 1, 1, 1),
id = "celltype",
ytree = FALSE,
cluster.order = celltype.order,
dot.col = c("#08519C", "white", "#A50F15")
)
# 注意指定自己喜欢的细胞类型顺序
averageHeatmap(
object = obj,
cluster.order = celltype.order,
gene.order = celltype_df$gene,
markerGene = celltype_df$gene
)
# 统计每个样本每个细胞类型的数量 分组bar图
CellStatPlot(
obj,
stat.by = "celltype",
group.by = "SampleID",
palcolor = dimCols
) + ggplot2::theme(aspect.ratio = 1 / 2)代码运行之后绘图结果:
如果遇到软件安装错误无法运行,可以使用组学大讲堂云服务器,已经安装好所有单细胞转录组数据分析的软件运行环境,省去环境配置麻烦,详情可扫下方二维码咨询:
Wang, T., Wang, F., Deng, S. et al. Single-cell transcriptomes reveal spatiotemporal heat stress response in maize roots. Nat Commun16, 177 (2025). https://doi.org/10.1038/s41467-024-55485-3
- 发表于 2026-04-08 15:04
- 阅读 ( 548 )
- 分类:转录组
