多组学联合分析—桑基图
做多组学研究的小伙伴,是不是常遇到这样的困扰?
转录组测出来上百个差异基因,代谢组筛出一堆差异代谢物,可这些“零散数据”怎么串联起来?基因调控了哪个通路?通路又如何影响代谢物变化?“基因-通路-代谢物”的调控链条藏在海量数据里,靠表格罗列根本看不清逻辑……
今天就给大家安利一款多组学整合分析的“可视化神器”——基因-通路-代谢物桑基图!它能把抽象的分子调控关系变成清晰的“流向图”,让你的科研逻辑一目了然,论文配图档次直接拉满!
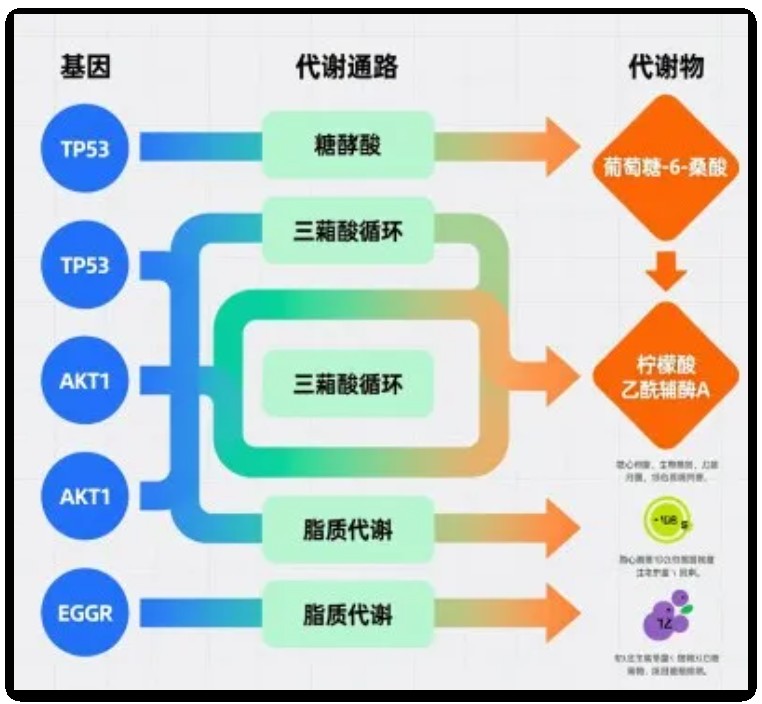
什么是基因-通路-代谢物桑基图
先给新手小伙伴科普下:桑基图原本是用来展示能量分流、物质流动的可视化工具,后来被广泛应用于生物信息学领域。而基因-通路-代谢物桑基图,就是专门为解析“基因调控通路、通路影响代谢物”这一核心逻辑设计的。
它的核心设计特别好理解,就3个关键点:
节点:图中的每个“小方块”,分别代表基因、通路、代谢物这三类“分子角色”,一眼就能区分不同层级的生物实体;
连线:连接节点的“线条”是核心!它代表不同角色之间的关联——比如基因指向通路,就说明这个基因参与了该通路的调控;通路指向代谢物,就代表通路激活/抑制会影响这类代谢物的合成或积累;
宽度:这是桑基图的“精髓”!连线的宽度可不是随便画的,而是对应了真实的实验数据——比如连线越宽,说明基因对通路的调控作用越强、通路与代谢物的关联越紧密。
下面我们使用R语言 ggsankey包教大家如何绘制桑基图,绘图脚本如下:
1.加载需要的包
#设置镜像,
local({r <- getOption("repos")
r["CRAN"] <- "http://mirrors.tuna.tsinghua.edu.cn/CRAN/"
options(repos=r)})
# 依赖包列表:自动加载并安装
package_list <- c("ggplot2","ggsankey","dplyr","tidyr","viridis")
# 判断R包加载是否成功来决定是否安装后再加载
for(p in package_list){
if(!suppressWarnings(suppressMessages(require(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))){
install.packages(p, warn.conflicts = FALSE)
suppressWarnings(suppressMessages(library(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))
}
}
2.读入数据
#设置工作路径
setwd("~/work/07.lh")
#读入数据
gene <- read.delim("gene.kegg.result.txt", sep = "\t", header = TRUE, stringsAsFactors = FALSE)
meta <- read.delim("meta.kegg.result.txt", sep = "\t", header = TRUE, stringsAsFactors = FALSE)
head(gene)
head(meta)
读入基因kegg富集分析结果和代谢物kegg富集分析结果,文件格式如下:
Pathway ID Pvalue RichFactor Sesquiterpenoid and triterpenoid biosynthesis TRINITY_DN31349_c0_g2 0.0000238520842.1058445728966 Sesquiterpenoid and triterpenoid biosynthesis TRINITY_DN4141_c1_g1 0.0000238520842.1058445728966 Sesquiterpenoid and triterpenoid biosynthesis TRINITY_DN4464_c0_g1 0.0000238520842.1058445728966 Sesquiterpenoid and triterpenoid biosynthesis TRINITY_DN588_c0_g2 0.0000238520842.1058445728966 Sesquiterpenoid and triterpenoid biosynthesis TRINITY_DN9758_c0_g1 0.0000238520842.1058445728966 PI3K-Akt signaling pathway TRINITY_DN2800_c0_g2 0.000742297381.21838150289017 PI3K-Akt signaling pathway TRINITY_DN2989_c0_g1 0.000742297381.21838150289017 PI3K-Akt signaling pathway TRINITY_DN6700_c0_g1 0.000742297381.21838150289017 PI3K-Akt signaling pathway TRINITY_DN874_c4_g1 0.000742297381.21838150289017 PI3K-Akt signaling pathway TRINITY_DN9367_c0_g2 0.000742297381.21838150289017
第一列:代谢通路;第二列:基因名称或代谢物名称;第三列:富集分析pvalue值;第四列:富集因子(该列可省略)
3.数据处理
# 为不同阶段指定不同的value
# 首先创建基因-通路连接的value(使用-log10(Pvalue))
gene_pathway <- gene %>%
distinct(ID, Pathway, .keep_all = TRUE) %>%
transmute(x = "Gene",
node = ID,
next_x = "Pathway",
next_node = Pathway,
value = -log10(Pvalue))
# 然后创建代谢物-通路连接的value(使用富集分数)
meta_pathway <- meta %>%
transmute(x = "Pathway",
node = Pathway,
next_x = "meta",
next_node = ID,
value = -log10(Pvalue))
# 然后创建代谢物 - NA 连接的value(使用富集分数)
meta_NA <- meta %>%
transmute(x = "meta",
node = ID,
next_x = "NA",
next_node = "NA",
value = -log10(Pvalue))
#pathway_NA
# 合并数据
df_sankey_custom_value <- bind_rows(gene_pathway, meta_pathway,meta_NA) %>%
mutate(fill_type = case_when(
x == "Gene" ~ "Gene",
x == "Pathway" ~ "Pathway",
x == "meta" ~ "meta",
))
df_sankey_custom_value[[1]] <- factor(df_sankey_custom_value[[1]], levels = c("Gene", "Pathway", "meta"))
df_sankey_custom_value[[3]] <- factor(df_sankey_custom_value[[3]], levels = c("Gene", "Pathway", "meta"))
head(df_sankey_custom_value)
4.绘图
ggplot(df_sankey_custom_value, aes(x = x,
next_x = next_x,
node = node,
next_node = next_node,
fill = factor(node),
label = node,
value = value)) + # 确保有value映射
#流线图层
geom_sankey(flow.alpha = 0.6, # 连接线的透明度(0-1)
node.color = "white", # 节点边框颜色
node.linewidth = 0.3, # 节点边框线宽
flow.color = "gray40", # 连接线边框颜色
smooth = 8, # 流线平滑度,值越大曲线越平滑
width = 0.2 ) + # 节点的相对宽度
#标签图层
geom_sankey_label(size = 2.5, # 标签字体大小
color = "black", # 标签文字颜色
fill = "white", # 标签背景填充色
alpha = 0.8, # 标签背景透明度
label.r = unit(0.05, "lines"), # 标签圆角
label.size = 0.2) + # 标签边框粗细
theme_sankey(base_size = 18) + # 桑基图专用主题,基础字体大小18
scale_fill_viridis_d() + # 使用viridis离散颜色标度
theme(
legend.position = "none",
plot.title = element_text(hjust = 0.5, face = "bold", size = 16),
plot.subtitle = element_text(hjust = 0.5, color = "gray40", size = 12),
axis.title.x = element_text(margin = margin(t = 10)),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.background = element_rect(fill = "white", color = NA),
panel.background = element_rect(fill = "white", color = NA)
) +
labs(
title = "桑基图",
x = "",
y = ""
)
结果如下图:
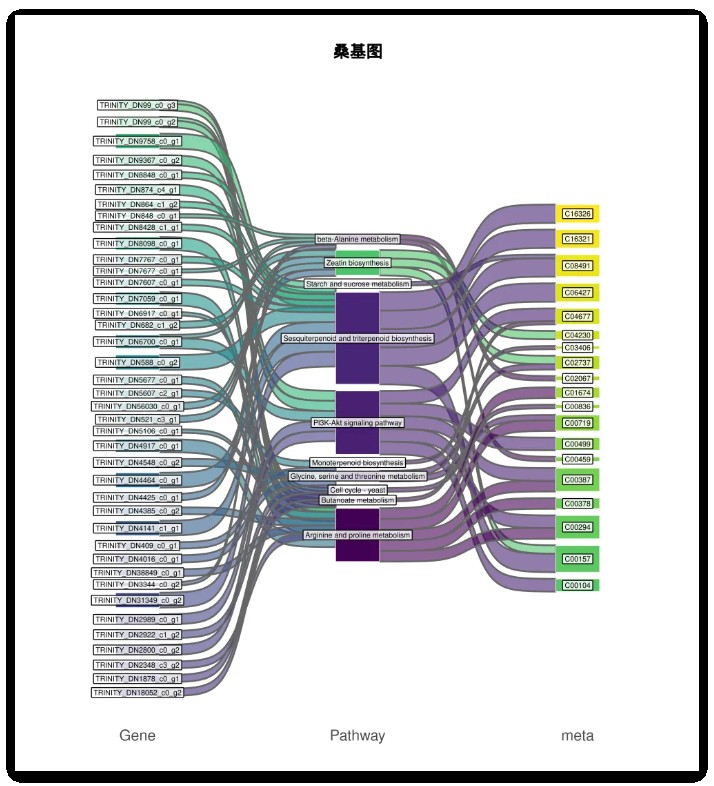
好了,小编就先给大家介绍到这里。希望对您的科研能有所帮助!祝您工作生活顺心快乐!
- 发表于 2026-01-05 17:15
- 阅读 ( 1507 )
- 分类:R
