一文教你绘制GO富集分析弦图,简单又好看
一文教你绘制GO富集分析弦图,简单又好看
一文教你绘制GO富集分析弦图,简单又好看
做生物信息学研究的小伙伴,是不是常被 “基因功能关联可视化” 难住?比如分析差异表达基因后,想直观展示基因与 GO 术语(生物过程、细胞组分、分子功能)的关联强度,普通柱状图、气泡图总差点意思 —— 要么看不清 “基因 - 术语” 的对应关系,要么体现不出富集程度的差异。
这时候,GO 富集分析弦图就能派上大用场!它像一张 “基因功能关系网”,用扇区代表基因和 GO 术语,用连线代表二者关联,还能通过颜色、粗细直观呈现富集显著性,让复杂的功能关联一目了然。
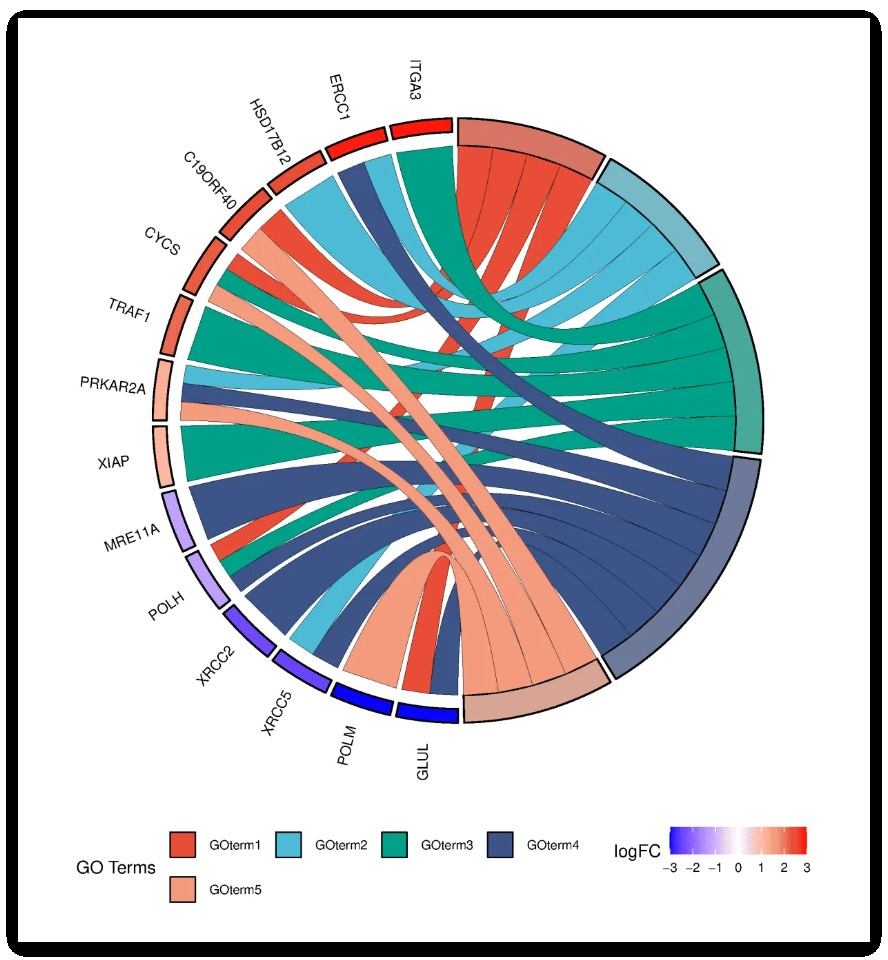
相较于传统的条形图或散点图,GO弦图的最大优势在于能同时展现多维信息:既能看到单个基因参与的多重功能,也能观察到特定功能术语涉及的基因集合。当面对复杂的基因表达数据时,这种全景式可视化有助于快速识别关键的生物学过程、发现潜在的功能协同效应,为后续实验验证提供清晰的研究方向。
今天就带大家从 “零” 开始,用最常用的 R 语言搞定弦图绘制,新手也能跟着做!
circlize包:专门画弦图的 “神器”。
# 1.安装CRAN来源常用包
#设置镜像,
local({r <- getOption("repos")
r["CRAN"] <- "http://mirrors.tuna.tsinghua.edu.cn/CRAN/"
options(repos=r)})
# 依赖包列表:自动加载并安装
package_list <- c("ggplot2","circlize","RColorBrewer","dplyr","tidyr")
# 判断R包加载是否成功来决定是否安装后再加载
for(p in package_list){
if(!suppressWarnings(suppressMessages(require(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))){
install.packages(p, warn.conflicts = FALSE)
suppressWarnings(suppressMessages(library(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))
}
}#设置工作路径
setwd("~/work/03.deg")
#读入数据
data <- read.delim("go.list.txt", sep = "\t", header = TRUE, stringsAsFactors = FALSE)
head(data)#GO_accession Description Corrected_P-value Rich_factor DEGgeneIDs GO:0097110 scaffold protein binding 0.00939529875.3061224489796 ENSG00000055118;ENSG00000006283 GO:0086091 regulation of heart rate by cardiac conduction 0.0221554599.5272727272727 ENSG00000006283;ENSG00000055118 GO:0035914 skeletal muscle cell differentiation 0.0276489763.1615384615385 ENSG00000005513;ENSG00000064300 GO:0031625 ubiquitin protein ligase binding 0.14923346713.0388692579505 ENSG00000055118;ENSG00000064300 GO:0005887 integral component of plasma membrane 0.17530195.28406881077038 ENSG00000040608;ENSG00000064300;ENSG00000002586;ENSG00000018280 GO:0032496 response to lipopolysaccharide 0.2171479818.1458563535912 ENSG00000018280;ENSG00000064300 GO:0007155 cell adhesion 0.2963382412.583908045977 ENSG00000002586;ENSG00000058085 GO:0006954 inflammatory response 0.2963382411.8570397111913 ENSG00000064300;ENSG00000018280 GO:0045944 positive regulation of transcription by RNA polymerase II 0.349636435.24664536741214 ENSG00000018280;ENSG00000005513;ENSG00000064195
# 获取所有唯一的基因ID
all_genes <- unique(unlist(strsplit(data$DEGgeneIDs, ";")))
# 获取所有唯一的Description
all_descriptions <- unique(data$Description)
# 创建空矩阵
pvalue_matrix <- matrix(0,
nrow = length(all_genes),
ncol = length(all_descriptions),
dimnames = list(all_genes, all_descriptions))
# 填充矩阵
for (i in 1:nrow(data)) {
genes <- strsplit(data$DEGgeneIDs[i], ";")[[1]]
description <- data$Description[i]
pvalue <- data$Corrected_P.value[i]
# 将p-value转换为-log10(p-value)
log_pvalue <- -log10(pvalue)
pvalue_matrix[genes, description] <- log_pvalue
}
# 查看结果矩阵
head(pvalue_matrix)
mat <- pvalue_matrix
# 过滤弱关联(可选)
mat[mat < 0.5] <- 0
# 只保留有连接的基因和GO术语
keep_genes <- rowSums(mat) > 0
keep_go <- colSums(mat) > 0
mat_filtered <- mat[keep_genes, keep_go, drop = FALSE]
# 获取基因和GO术语名称
gene_ids <- rownames(mat_filtered)
go_terms <- colnames(mat_filtered)# 设置颜色方案
# 基因统一颜色
mycolor_palette <-c('#E5D2DD', '#53A85F', '#F1BB72', '#F3B1A0', '#D6E7A3', '#57C3F3', '#476D87',
'#E59CC4', '#AB3282', '#23452F', '#BD956A',
'#9FA3A8', '#E0D4CA', '#5F3D69', '#58A4C3', '#E4C755', '#F7F398',
'#AA9A59', '#E63863', '#E39A35', '#C1E6F3', '#6778AE', '#91D0BE', '#B53E2B',
'#712820', '#DCC1DD', '#CCE0F5', '#CCC9E6', '#625D9E', '#68A180', '#3A6963',
'#968175','#585658')
#scales::show_col(mycolor_palette,cex_label = 0.5) #显示一下颜色
#gene_color <- rep("lightblue", length(gene_ids))
gene_color <- rep("#3C77AF", length(gene_ids))
gene_color <- rep("#6CB8D2", length(gene_ids))
#gene_color <- rep("#4991C1", length(gene_ids))
names(gene_color) <- gene_ids
# GO术语不同颜色(使用彩虹色系)
#go_colors <- rainbow(length(go_terms))
go_colors <- head(mycolor_palette, length(go_terms))
names(go_colors) <- go_terms
# 设置网格颜色:基因统一颜色,GO术语各自颜色
#grid_colors <- c(go_colors,rep(gene_color, length(gene_ids)))
grid_colors <- c(go_colors,gene_color)
head(grid_colors)
# 设置弦的颜色:与目标GO术语颜色一致
# 对于每个连接,使用GO术语的颜色
link_colors <- matrix(NA, nrow = nrow(mat_filtered), ncol = ncol(mat_filtered))
for(i in 1:nrow(mat_filtered)) {
for(j in 1:ncol(mat_filtered)) {
if(mat_filtered[i, j] > 0) {
link_colors[i, j] <- go_colors[j] # 弦颜色与GO术语颜色一致
}
}
}
# 设置顺序:先基因,后GO术语
order_vector <- c( go_terms,gene_ids)
# 清除之前的圆形布局
circos.clear()
# 设置左右布局
n_genes <- length(gene_ids)
n_go <- length(go_terms)
gap_degrees <- c(rep(2, n_genes - 1), 20, rep(2, n_go - 1), 20)
#circos.par(start.degree = 90, gap.degree = gap_degrees)
# 在绘制弦图前设置边距
circos.par(
start.degree = 90, # 起始角度(影响布局)
gap.degree = gap_degrees, # 设置不同组间的间隔
track.margin = c(0.01, 0.01), # 轨道边距(上下)
canvas.xlim = c(-1.2, 1.2), # 画布X轴范围
canvas.ylim = c(-1.2, 1.2) # 画布Y轴范围
)
# 绘制弦图
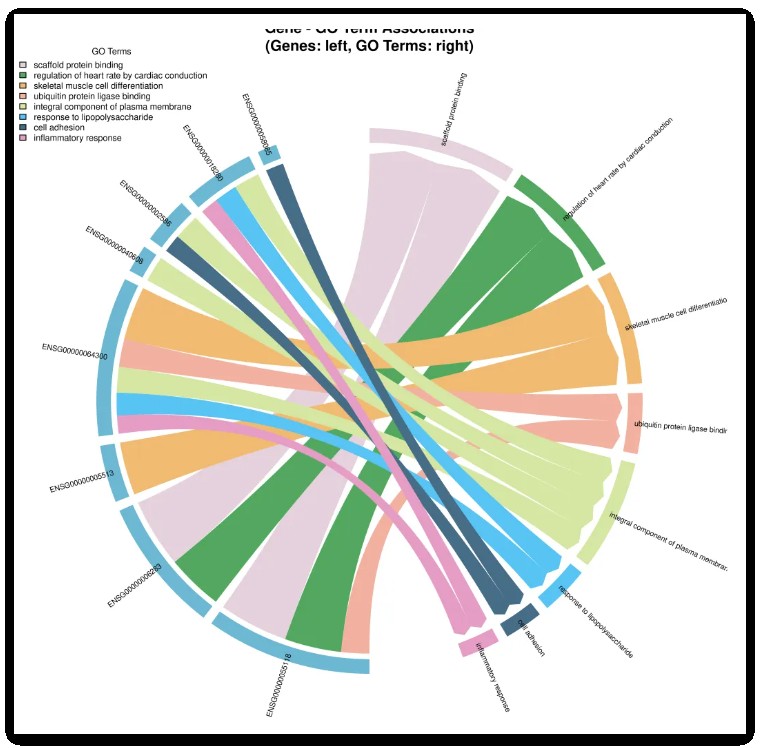
chordDiagram(mat_filtered,
order = order_vector,
grid.col = grid_colors, # 基因统一颜色,GO术语不同颜色
col = link_colors, # 弦颜色与GO术语一致
transparency = 0.5,
directional = 1,
direction.type = "arrows",
link.arr.type = "big.arrow",
annotationTrack = "grid", # 只显示网格
annotationTrackHeight = mm_h(5))
# 添加标签
circos.track(track.index = 1, panel.fun = function(x, y) {
sector.index <- get.cell.meta.data("sector.index")
xlim <- get.cell.meta.data("xlim")
ylim <- get.cell.meta.data("ylim")
circos.text(mean(xlim), mean(ylim), sector.index,
facing = "clockwise",
niceFacing = TRUE,
#adj = c(0.5, 0.5),
adj = c(0, 0.5), #基因名称放在圈外
cex = 0.6)
}, bg.border = NA)
# 添加图例
legend("topleft",
legend = go_terms,
fill = go_colors,
title = "GO Terms",
bty = "n",
cex = 0.7,
ncol = 1)
title("Gene - GO Term Associations\n(Genes: left, GO Terms: right)")结果如下图:
好了,小编就先给大家介绍到这里。希望对您的科研能有所帮助!祝您工作生活顺心快乐!
- 发表于 2026-01-05 17:08
- 阅读 ( 1733 )
- 分类:R
