怎么得到基因组之间的结构变异结果?
在泛基因组研究中,结构变异(SVs)的检测是揭示物种内遗传多样性的关键环节。相比单核苷酸多态性(SNPs),结构变异能够解释更多的表型差异和适应性特征。今天我们就来深入探讨泛基因组分析中...
在泛基因组研究中,结构变异(SVs)的检测是揭示物种内遗传多样性的关键环节。相比单核苷酸多态性(SNPs),结构变异能够解释更多的表型差异和适应性特征。今天我们就来深入探讨泛基因组分析中结构变异的完整分析流程。
数据准备与标准化
在进行比较之前,确保所有基因组的染色体命名一致:
# 使用sed统一染色体命名
sed 's/>chr/>Chr/g' genome1.fa > genome1_standard.fa
注意事项:确保比较的基因组版本一致,染色体数量和质量可控。
基因组比对
1.1 同种基因组比较(使用MUMmer)
# 生成全局比对
nucmer --maxmatch -g 1000 -c 90 -l 40 -t 10 -p 前缀 ref.fa genome1_standard.fa
1.2 同属不同种基因组比较(使用minimap2)
# 长序列比对
minimap2 -x asm5 -t 24 -c ref.fa genome1_standard.fa > 前缀.paf
# -x asm5:基因组间比对预设参数
# -t 24:使用8线程
# -c:输出CIGAR字符串
# 基于ragtag进行格式转换
ragtag_paf2delta.py -r ref.fa -q genome1_standard.fa 前缀.paf > 前缀.delta
2. 过滤比对结果
delta-filter -r -q -i 90 -l 100 前缀.delta > 前缀.1delta
#-q:查询序列锚定
# r:参考序列锚定
# -l 最小比对长度
# -i 最小比对率
# 上面没写但是实际会用到的:-1 1对1的比对结果;-m 多对多的比对结果
SyRI进行结构变异检测
1. 将比对结果转换为SyRI输入格式:
show-coords -Tclrd 前缀.1delta > alignment.coords
2. 运行SyRI:
syri -c alignment.coords -d 前缀.1delta -r ref.fa -q genome1_standard.fa --nc 24 --prefix 前缀
# -c .coords文件
# -d delta文件
# -r 参考基因组序列所在位置
# -q 查询序列所在位置
# -nc 线程数
# --prefix 输出结果前缀
输出包括:SNPs、插入缺失、倒位、易位等变异;复制区域和重复序列信息;基因组共线性区块
3. plotsr 绘图:
# 整理绘图config文件
echo -e "##ft = File type (fa:fasta/cl:chromosome_length, default = fa. cl files are fasters)
##lc = line colour
##lw = line width
##file name tags
ref.fa\t参考基因组名字\tft:fa;lw:1.5
genome1_standard.fa\t查询基因组名字\tft:fa;lw:1.5" > genome.txt
###绘图
plotsr --sr 前缀syri.out --genomes genome.txt -o plotsr.pdf
4. 结果:
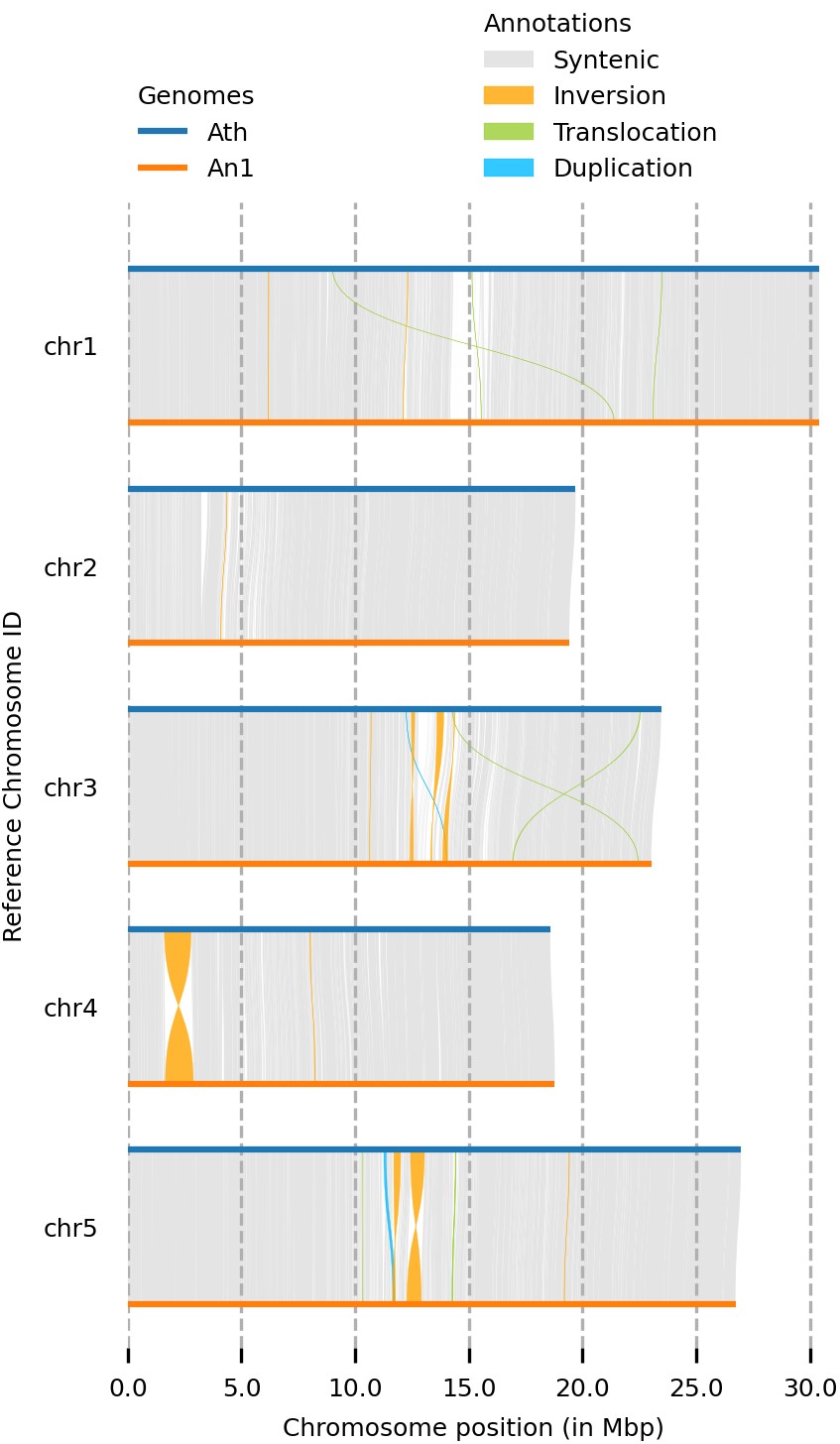
变异检测与图形泛基因组构建
获得高质量结构变异后,即可构建泛基因组
1. 变异vcf合并
将每个样本的SVs利用SURVIVOR合并后得到一个涵盖所有样本结构变异的vcf文件。
2.泛基因组图谱构建
基于参考基因组和变异信息的构建方式,在vg软件中输入合并之后的sv-vcf文件和参考基因组序列文件,即可构建出泛基因组文件。
vg construct -r ref.fa -v sv.vcf.gz -a -t 12 -m 100 > pangenome.vg
## -m, --node-max N
## -t, --threads N
## -r, --reference FILE
## -v, --vcf FILE
## -a, --alt-paths
参考:
https://bdtcd.xetslk.com/s/lIqL9
- 发表于 2025-10-10 16:11
- 阅读 ( 2242 )
- 分类:基因组学
