相关性分析及热图绘制-脚本使用
相关性分析是指对两个或多个具备相关性的变量进行分析,从而衡量变量间的相关程度。变量之间相关程度的量化使用相关系数r,常见的相关系数有Pearson相关系数、Spearman相关系数以及Kendall相关系数。生信分析中常绘制热图直观清晰地了解两两变量间的相关性。
在2021年发表于The Journal for ImmunoTherapy of Cancer上的影响因子为13.75的文章中,作者进行了相关性分析并绘制了热图,直观清晰地展现了两两变量间的相关情况。
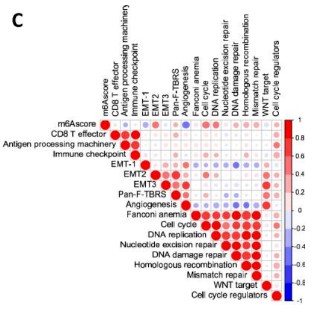
 下图图C就是作者在m6Ascore值、已知生物学过程富集评分的两两之间进行相关性分析并对相关系数r绘制热图。其中蓝色的点表示负相关关系,红色的点表示正相关关系,点的大小以及颜色的深浅都表示相关系数的大小。
下图图C就是作者在m6Ascore值、已知生物学过程富集评分的两两之间进行相关性分析并对相关系数r绘制热图。其中蓝色的点表示负相关关系,红色的点表示正相关关系,点的大小以及颜色的深浅都表示相关系数的大小。
为了使大家能更简便快捷地进行相关性分析并绘制出精美的相关性热图,这里我们给大家提供两个R脚本,一个脚本用于进行相关性分析,另一个脚本用于对相关性分析结果进行筛选并绘制相关性热图。这两个脚本都只需要准备好相应的输入文件,再进行简单的命令行操作即可。
使用命令示例如下
#相关性分析
Rscript $scriptdir/corr.r -l DDR_deg_gene_TPM.tsv -g DDR_deg_gene_TPM.tsv -p DDR_deg
上述命令得到两个结果文件,分别为显著性p值(DDR_deg_corr_p.tsv):
| BRCA1 | RAD51 | RAD54L | |
| BRCA1 | 0 | 1.8046795897094e-56 | 4.32081702742339e-82 |
| RAD51 | 1.8046795897094e-56 | 0 | 7.56127779085093e-70 |
| RAD54L | 4.32081702742339e-82 | 7.56127779085093e-70 | 0 |
| BRCA1 | RAD51 | RAD54L | |
| BRCA1 | 1 | 0.624364338753274 | 0.718352221206303 |
| RAD51 | 0.624364338753274 | 1 | 0.677749484949762 |
| RAD54L | 0.718352221206303 | 0.677749484949762 | 1 |
#绘制热图时不对相关性分析结果进行筛选
Rscript $scriptdir/corrplot_select.r -p DDR_deg_corr_p.tsv -r DDR_deg_corr_r.tsv \
--corrplot -t upper -d AOE -f test
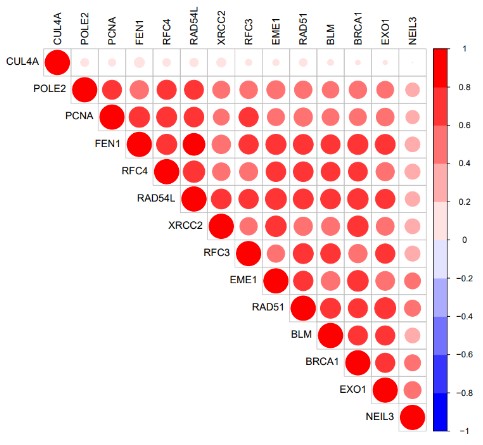
#以p<0.001,r>0.4筛选相关性分析结果,此时不绘制热图
Rscript $scriptdir/corrplot_select.r -p DDR_deg_corr_p.tsv -r DDR_deg_corr_r.tsv \
-P 0.001 -R 0.4 -f test
| ID |
| BRCA1 |
| RAD51 |
RAD54L |
输入文件准备
| ID | TCGA-91-6840-01A-11R-1949-07 | TCGA-78-8655-01A-11R-2403-07 | TCGA-NJ-A7XG-01A-12R-A39D-07 |
| BRCA1 | 7.69213640867971 | 2.49968743079702 | 2.20722489973144 |
| RAD51 | 12.3094747327104 | 4.51457829240412 | 2.11795151239397 |
| RAD54L | 9.32184896562367 | 4.58951017219995 | 1.36208162672449 |
更多脚本参数设置及说明
初次使用脚本时,可以通过-h参数获得以下帮助信息。
Rscript $scriptdir/corr.r -h
usage: /share/work/fangs/scripts/tcga/corr.r [-h] -l lnc_data -g gene_data
[-m method] [--filter] [-e exp]
[-n num] [-o outdir] [-p prefix]
corr_plot
optional arguments:
-h, --help show this help message and exit
-l lnc_data, --lnc_data lnc_data
input data file path[required]
-g gene_data, --gene_data gene_data
input data file path[required]
-m method, --method method
input the type of method that calculates the
correlation[optional,default pearson]
--filter Whether to filter low expression
genes[optional,default:False]
-e exp, --exp exp Minimum gene expression during
filtration[optional,default:1]
-n num, --num num The number of samples that meet the gene expression
level during filtration,A value of 2 means that at
least half of the samples meet the criteria,and so
on[optional,default:2]
-o outdir, --outdir outdir
output file directory[optional,default cwd]
-p prefix, --prefix prefix
out file name prefix[optional,default specified-
gene_lncRNAs]Rscript $scriptdir/corrplot_select.r -h
usage: /share/work/fangs/scripts/tcga/corrplot_select.r [-h] -p P_DATA -r
R_DATA [-P P_VALUE]
[-R R_VALUE]
[--corrplot] [--add_r]
[-t TYPE] [-d ORDER]
[--Rdata] [-H HEIGHT]
[-W WIDTH] [-o OUTDIR]
[-f PREFIX]
Ferroptosis-Related lncRNAs
optional arguments:
-h, --help show this help message and exit
-p P_DATA, --p_data P_DATA
input data file path[required]
-r R_DATA, --r_data R_DATA
input data file path[required]
-P P_VALUE, --P_value P_VALUE
P value of correlation[optional,default 0.05]
-R R_VALUE, --R_value R_VALUE
the correlation coefficient[optional,default 0.3]
--corrplot Whether to draw a correlation heat
map[optional,default:False]
--add_r Whether to show correlation coefficients in the
graph[optional,default:False]
-t TYPE, --type TYPE Specifies the presentation style, which can be
'full','lower' or 'upper'[optional,default upper]
-d ORDER, --order ORDER
Specify how correlation coefficients are arranged,
which can be 'AOE','FPC' or 'hclust'[optional,default
AOE]
--Rdata Whether to output filtered correlation coefficient
matrix[optional,default:False]
-H HEIGHT, --height HEIGHT
the height of dumbbell plot[optional,default 8]
-W WIDTH, --width WIDTH
the width of dumbbell plot[optional,default 8]
-o OUTDIR, --outdir OUTDIR
output file directory[optional,default cwd]
-f PREFIX, --prefix PREFIX
out file name prefix[optional,default fer_lnc]第一个脚本corr.r:-m 指定进行相关性分析所用的方法,默认为皮尔逊相关性分析;
--filter 当输入文件为基因表达量时,指定是否过滤低表达的基因,在命令中添加该参数即为要进行过滤;
-e 指定过滤时基因在样本中所要达到的最低表达量,默认为1;
-n 默认为2,当-e、-n参数都为默认值时,即筛选至少有一半样本表达大于1的基因,-n指定为3,即筛选至少有三分之一样本表达大于1的基因,以此类推。
第二个脚本corrplot_select.r:
-P 指定筛选结果时显著性p值的阈值,默认为0.05;
-R 指定筛选结果时相关系数r的阈值,默认为0.3;
--corrplot 指定是否绘制相关性热图,在命令中添加该参数即为要进行绘制;
--add_r 指定是否在绘制的相关性热图中显示相关系数r;
-t 指定热图展示的形状,默认为上三角形;
-d 指定热图中相关系数r的排列方式,默认为AOE;
--Rdata 指定是否输出筛选后的相关系数r值结果,在命令中添加该参数即为输出。
其他参数说明
-H 指定输出图片的高度,默认高为6英寸;
-W 指定输出图片的宽度,默认宽为8英寸;
-o 指定结果输出的路径,默认为当前路径;
-p 指定输出文件名字的前缀。
脚本获取方法
为了方便感兴趣的学员小伙伴想复现这个分析内容,我把数据和代码一并打包,免费供大家使用学习。只需要如下几步,即可获得下载链接。
1、关注生信博士公众号;
2、转发本文章至朋友圈,并截图;
3、在公众号菜单栏回复朋友圈截图和corr即可获得下载链接。
如果没有数据分析的基础,不知道从何下手,可以学习一下我们的《医学数据分析环境搭建》视频课程,扫描下方二维码了解课程详情。

- 发表于 2022-06-10 15:04
- 阅读 ( 10665 )
- 分类:R
