SNP密度图
我们做分析的时候经常会需要统计snp/INDEL/变异密度的信息,这个时候有以下几种选择
一、vcftools统计密度,而后绘图
二、CMplot包直接绘图
其中二明显步骤更少,所以在这里简单给大家介绍一...
我们做分析的时候经常会需要统计snp/INDEL/变异密度的信息,这个时候有以下几种选择
一、vcftools统计密度,而后绘图
二、CMplot包直接绘图
其中二明显步骤更少,所以在这里简单给大家介绍一下怎么做,有时间再重新编辑这个文章补充1的做法和代码
CMplot包直接绘图
1. linux:vcf转化为map格式
plink --vcf input.vcf --recode --out output --allow-extra-chr --set-missing-var-ids @:#
这样就可以得到output为前缀的ped和map格式文件。这里呢vcf.gz也可以直接作为-vcf的输入文件,可能plink会出现Segmentation fault (core dumped),但是看了一下没有影响结果。
其中map格式是我们想要继续使用的,第一列是染色体;第二列是变异的ID,这里因为我们vcf里面其实没有ID,所以根据我们的指定--set-missing-var-ids @:#创建了ID;第四列是变异所在位置:
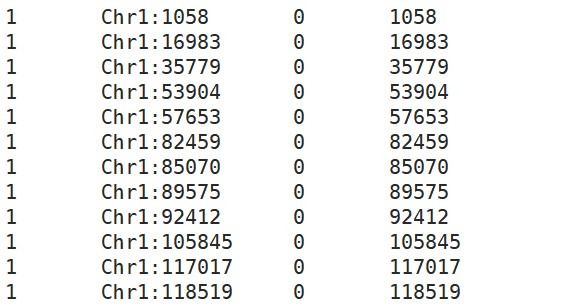
2. R:整理输入数据
setwd("/workdir")
d <- read.table("output.map",header=F)
d <- d[d$V1==1, c(2,1,4)] #我这里面就选择了一条染色体画图,大家可以选择全部绘制
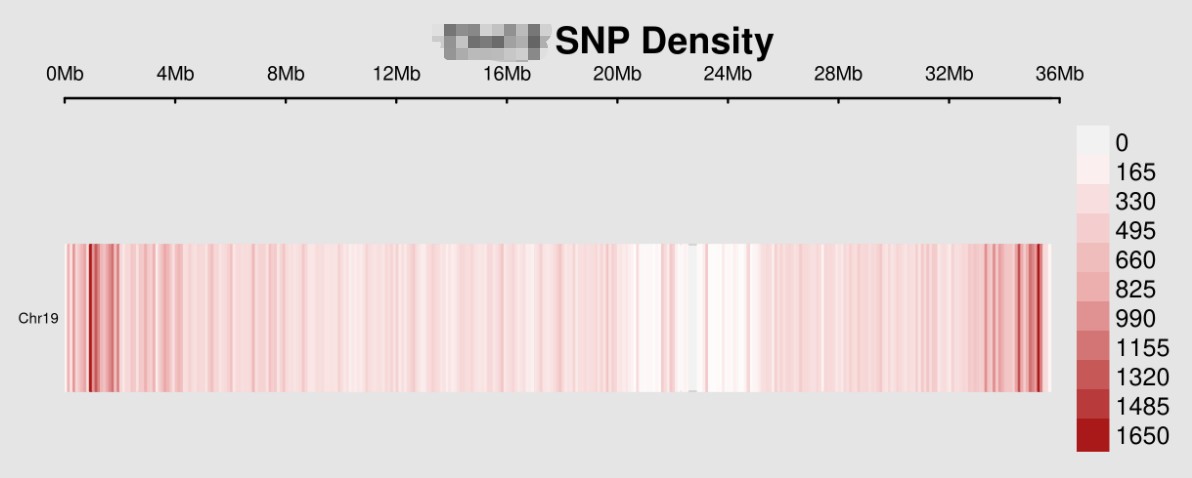
3. R:CMplot绘图
CMplot(d,
plot.type = "d", # 绘制密度图,指定为d
bin.size = 1e5, # 指定求bin的bin大小,热图本质上就是一列一列的有颜色的格子排起来,这就是在指定每一列的物理距离是多少
chr.den.col = c("white", "#E2B0B0", "#992727"), # 指定绘图颜色,默认是绿色、黄色到红色
file = "pdf", #指定输出文件格式
file.name = "SNP_density", # 指定输出文件名称,其实这里指定的是中缀
dpi = 300,
main = "SNP Density", # 图上面的标题
file.output = TRUE,
verbose = TRUE,
width = 9,
height = 4)
出来的结果是这样:
4.R绘图优化
如果我们有多个数据都要画图最后要拼在一起,这个时候比例尺不一样怎么办?
有一个参数可以解决这个问题: bin.rang=c(0,1000)
只要我们制定以下这个bin的统计范围,就可以让颜色梯度统一起来。
- 发表于 2026-03-19 11:21
- 阅读 ( 787 )
- 分类:R
