ggbio绘制基因结构图
基因结构图是基因组学研究中最基础的可视化之一,它能帮助我们直观展示基因的外显子-内含子结构;定位突变位点、SNP位点在基因中的位置;展示不同转录本的结构差异。ggbio是基于ggplot2开发的基...
基因结构图是基因组学研究中最基础的可视化之一,它能帮助我们直观展示基因的外显子-内含子结构;定位突变位点、SNP位点在基因中的位置;展示不同转录本的结构差异。ggbio是基于ggplot2开发的基因组数据可视化专精包,特别适合处理基因组坐标数据。它够直接处理GRanges对象(基因组范围对象),与Bioconductor生态系统完美契合。
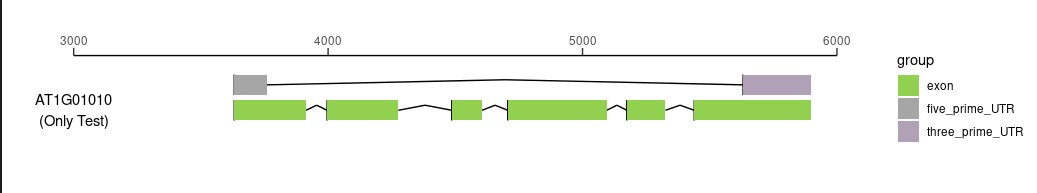
先来看看最终效果!
R包安装
通常来说直接install.packages()就可以直接安装了,但是有的时候可能报错版本问题,这个时候可以借助BiocManager或者是devtools安装R包
install.packages(c("ggplot2", "ggh4x"))
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("ggbio", "GenomicRanges"))
数据准备
需要准备gff文件,保留自己想要展示的元件及其位置信息即可。
存在overlap关系的元件需要进行取舍。
原始数据:
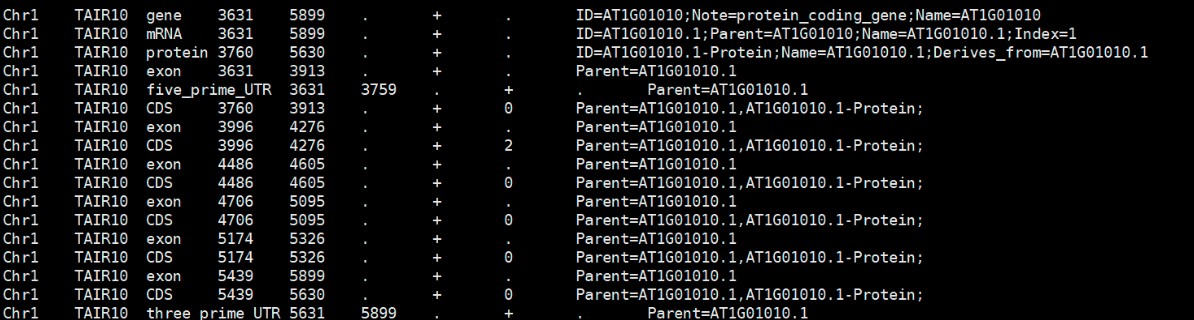
操作:
# 选择展示exon和UTR区
grep "AT1G01010" TAIR10_GFF3_genes.gff | awk '$3!~/[gNCDS]+/' | grep -v "protein" > test.gff
之后的数据:
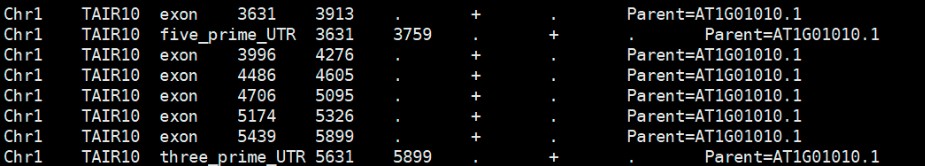
图像绘制
1. 载入R包:
library(ggh4x)
library(ggplot2)
library(ggbio)
library(GenomicRanges)
2. 读入数据:
#赋值变量,根据自己绘图数据来。gene是gff前缀的名称
gene <- "test"
chr <- "Chr1"
start <- 3000
end <- 6000
df <- read.csv(paste0(gene,".gff"),header = F,sep = "\t")
head(df)
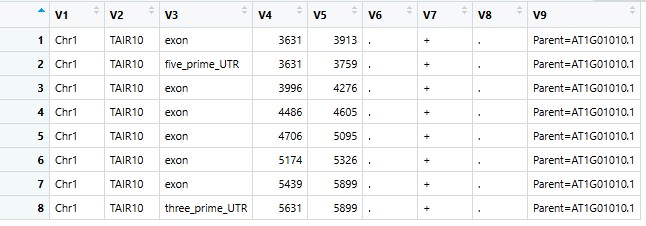
3. 绘图:
# 整理成绘图数据
pos <-GRanges(chr,ranges=IRanges(start=df$V4,end=df$V5,group=df$V3))
# 绘图
autoplot(pos,aes(fill=group),geom="alignment")+
theme_bw()+
scale_x_continuous(limits=c(start,end),
breaks =c(seq(start,end,by=1000)),
position ="top")+
theme(panel.border =element_blank(),
panel.grid =element_blank(),
axis.line.x=element_line(),
axis.ticks.length = unit(0.2,'cm'))+
guides(x=guide_axis_truncated(trunc_lower = start,
trunc_upper = end))+
theme(aspect.ratio =0.1)+
scale_y_continuous(limits=c(0,3))+
theme(axis.text.y=element_blank(),
axis.ticks.y=element_blank())+
annotate(geom ="text",x=start,y=1,
label="AT1G01010\n(Only Test)")+
scale_fill_manual(values = c("#92d050","#a6a6a6","#b0a1b7"))+
coord_cartesian(clip="off")
4. 结果:
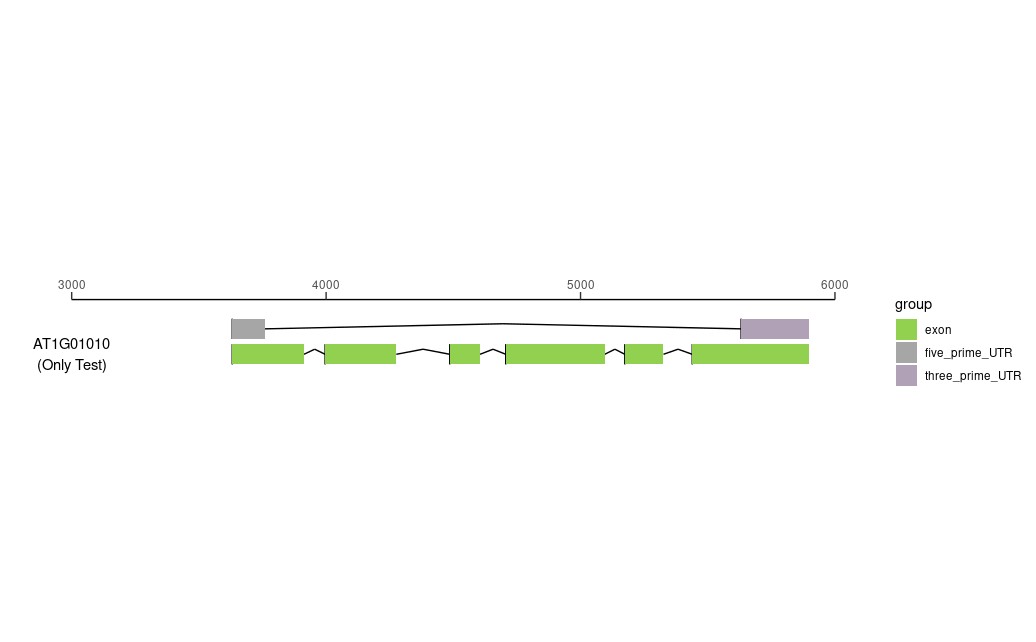
绘图大小由“scale_x_continuous”指定,坐标轴由“guides”规定。
“annotate”规定了左侧的基因ID。
不同区域颜色由“scale_fill_manual”指定,而能指定颜色的根本原因在于绘图开始的“aes(fill=group)”,这里面的group呼应整理绘图数据这一步的pos。
鉴于UTR区域和exon存在重合所以分成两个轨迹展示了,如果在这里选择用CDS和UTR做展示就不会出现两个轨迹。
参考:
https://zhuanlan.zhihu.com/p/380914002
- 发表于 2025-09-09 15:44
- 阅读 ( 3562 )
- 分类:R
