gff文件格式不标准的修改脚本
gff文件第三列一般有gene,mRNA,CDS,exon等等信息,但是有时候我们下载的gff文件没有gene信息,只有mRNA等等的信息,我们可以实用脚本手动校正。
出现问题
gff文件第三列一般有gene,mRNA,CDS,exon等等信息,但是有时候我们下载的gff文件没有gene信息,只有mRNA等等的信息,比如下图:
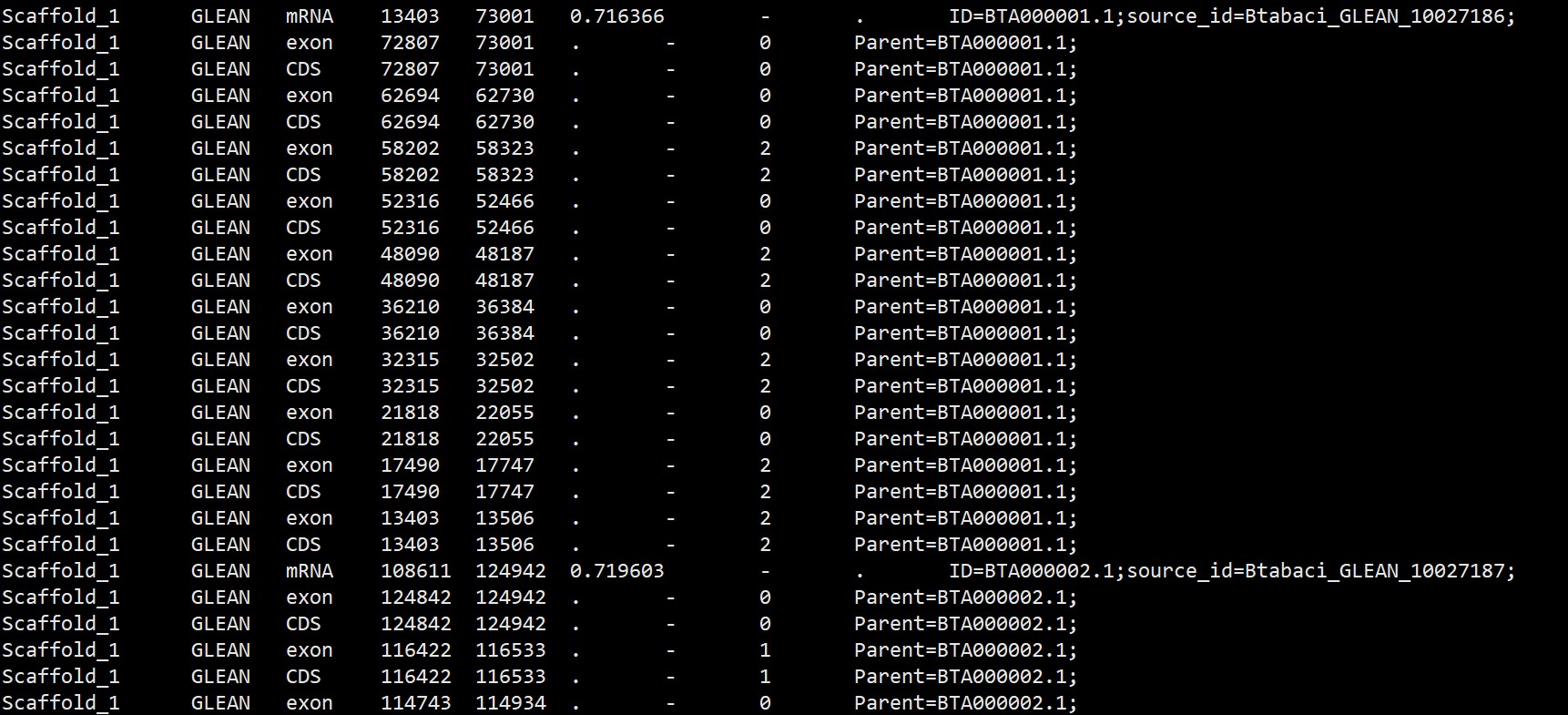
之前有用使用awk更改的回答,用awk命令批量添加gene行,把mRNA的ID作为基因ID,并且在mRNA行添加Parent信息:参考:GFF文件格式不标准,第三列只有mRNA处理方法 - 组学大讲堂问答社区 (omicsclass.com)
但这样更改之后,gene的ID会和mRNA一致,mRNA的ID和Parent信息也会相同,比如下方信息,这样的话在一些分析中可能就会报错,我们需要将他改成正确的格式
#gene的ID会和mRNA一致,mRNA的ID和Parent信息也会相同,可能会出错
Scaffold_1 GLEAN gene 13403 73001 0.716366 - . ID=BTA000001.1;
Scaffold_1 GLEAN mRNA 13403 73001 0.716366 - . ID=BTA000001.1;Parent=BTA000001.1;
#正确
Scaffold_1 GLEAN gene 13403 73001 0.716366 - . ID=BTA000001;
Scaffold_1 GLEAN mRNA 13403 73001 0.716366 - . ID=BTA000001.1;Parent=BTA000001;
代码思路
我们可以对其人为修改,我们需要
1.批量添加gene行,把mRNA的ID作为基因ID,同时去掉ID属性中的.1或.2或.3等这种.加数字的组合;
2.在mRNA行添加Parent信息,Parent信息为gene的ID信息,并把最后一列的source_id=Btabaci_GLEAN_10027186;类似的属性删掉,即mRNA行最后一列只有ID信息和Parent 信息;
3.gene行位于每个mRNA的上一行。
代码
以下为Python脚本,使用方式为 python gff.py >new.gff
需要提前将with open('input.gff', 'r') as f: 中的inpu.gff 改成自己的该文件名字
import re
# 读入文件
with open('input.gff', 'r') as f:
lines = f.readlines()
# 初始化基因ID
gene_id = ''
# 遍历每一行
for line in lines:
if line.startswith('##'):
# 忽略以##开头的注释行
continue
elif line.startswith('#'):
# 对于以#开头的注释行,直接输出
print(line.strip())
else:
# 对于数据行
fields = line.strip().split('\t')
# 如果是mRNA行
if fields[2] == 'mRNA':
# 获取mRNA ID
mrna_id = re.findall(r'ID=([^;]+)', fields[8])[0]
# 生成新的基因ID
gene_id = re.sub(r'\.\d+', '', mrna_id)
# 添加gene行
gene_line = [fields[0], fields[1], 'gene', fields[3], fields[4], fields[5], fields[6], fields[7], f'ID={gene_id};']
print('\t'.join(gene_line))
# 修改mRNA行
mrna_line = fields
mrna_line[2] = 'mRNA'
mrna_line[8] = f'ID={mrna_id};Parent={gene_id};'
mrna_line[8] = re.sub(r';source_id=[^;]+', '', mrna_line[8])
print('\t'.join(mrna_line))
else:
# 对于其他行直接输出
print(line.strip())
这个脚本可以完成如下任务:
1.读取输入文件(gff文件);
2.对于每一行,根据第3列的内容(gene/mRNA/CDS/exon等),分别进行处理:
对于gene行,直接输出;
对于mRNA行,根据ID属性值生成gene行的ID属性值,并输出gene行和修改后的mRNA行;
对于其他行(CDS/exon等),根据Parent属性值生成修改后的行,并输出;
3.将处理后的结果写入输出文件。
这个脚本的重难点和知识点如下:
- 正则表达式的使用:这个脚本使用了多个正则表达式来匹配和替换不同的模式。正则表达式是一个非常强大的工具,但是对于一些人来说可能比较难以理解和掌握。
- 字符串操作:这个脚本涉及到了多个字符串的操作,包括分割、替换、拼接等等。对于字符串操作不熟悉的人来说,可能需要花费更多的时间来理解和编写这些代码。
- GFF格式的解析:这个脚本需要对GFF格式的文件进行解析和处理,需要了解GFF格式的规范和约定,以及相关的属性和标签的含义。
- 编程思维:最后,这个脚本需要有一定的编程思维,能够将需求转化为代码实现的思路和能力。这可能需要一些经验和实践才能掌握。
- 发表于 2023-04-07 16:02
- 阅读 ( 5070 )
- 分类:python
