孟德尔随机化(MR)
前面的文章中我们给大家介绍过eQTL和sQTL的概念, 大体总结一下,eQTL是指基因组上的一些突变可以改变相关基因的表达,sQTL是指这些突变同时也会导致可变剪接的差异,实际上这两个都是归于一个叫做分子定量性状基因座(molQTL或者xQTL)的大家族里,为了确定GWAS识别的与复杂性状或疾病相关的遗传变异位点是否与xQTL共享相同的因果变异,还可以使用共定位分析来推断遗传变异位点是否通过调控基因表达或其他分子表型(如剪接、甲基化)来影响性状
那么在此基础上,给大家引入一个新的概念——孟德尔随机化
(Mendelian Randomization, MR)
孟德尔随机化是一种利用遗传变异作为工具变量(IV)的因果推断方法,模拟随机对照试验,来估计暴露因素(如xqtl)对结局性状(如疾病风险)的因果效应。它避免了传统观察性研究的混杂和逆因果偏倚,核心依赖孟德尔遗传定律(等位基因随机分配)
计算逻辑:
- 核心假设验证:检查三个IV(独立snp)假设——IV与暴露强相关(用F统计量评估);IV不直接影响结局以外路径;IV仅通过暴露影响结局。
- 效应估计:对于每个IV,计算其对暴露的效应除以对结局的效应,得到单个因果估计。然后,通过加权平均(如逆方差加权)合并所有IV的估计,权重基于结局效应的精确度(标准误倒数平方)。这产生整体因果效应值(如beta系数或OR)。
- 敏感性分析:用多种方法验证鲁棒性——一种方法假设所有IV有效,直接平均;另一种检测截距是否偏离零来识别多效性并校正;中位数方法对半数以上有效IV鲁棒;异常检测方法识别并移除离群SNP。异质性测试检查IV间一致性,如果高则提示无效IV。
- 结果解读:如果主分析和敏感性一致,且假设满足,则支持因果;否则需调整IV或报告偏倚。适用于两样本(暴露和结局GWAS不同人群)以减少样本重叠偏倚。
和共定位的关系:
共定位是MR的前提验证, MR假设无水平多效性,但GWAS信号常因LD混杂。共定位先确认“暴露xQTL与结局GWAS共享同一因果SNP”,再用该SNP作为强IV做MR,避免多效性偏倚,他们的关系是互补而非替代
MR软件
这里我们介绍SMR这个软件, 他严格来说并非是最标准定义的孟德尔随机化,但却是最泛用的,有着很强的联合分析潜力,所以被广泛应用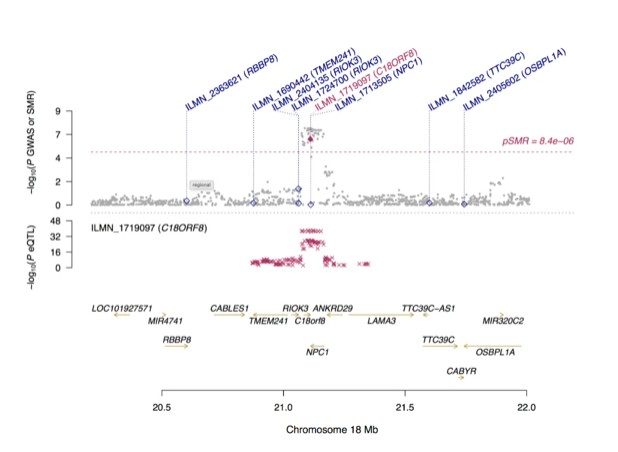
染色体18上基因表达与某个表型(疾病/性状)的因果关联分析1.SMR
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM & Yang J (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets
https://www.nature.com/articles/ng.3538
SMR是一种基于总结数据的孟德尔随机化变体,专门整合eQTL(基因表达定量位点)和GWAS数据,测试特定基因表达水平对复杂性状的因果影响。它高效处理大规模公共数据,优先使用eQTL中最强关联SNP作为工具变量,同时内置测试区分真正因果与LD污染

安装很简单,clone以后直接make就可以,见下面的代码
git clone https://github.com/JianYang-Lab/SMR.gitcd SMR && mkdir build && cd buildcmake ..make
网站内有文字教程,关于一些用法,链接如下
https://yanglab.westlake.edu.cn/software/smr/#SMR&HEIDIanalysis
以基础分析为例:
smr \ --bfile mydata \ #plink2格式的SNP数据 --gwas-summary gwas.ma \ #输入的gwas数据 --beqtl-summary xqtl \ #输入的xqtl数据(bsed格式) --out smr \ #输出文件 --thread-num 10 #线程数
基因型数据可以提供plink2或者bld格式(这个可以直接smr自己获取,使用基因型计算的LD信息,使用--bld替换--bfile参数,,不熟悉的直接忽略,只用plink2就行)
gwas文件格式
SNP A1 A2 freq b se p nrs1001 A G 0.8493 0.0024 0.0055 0.6653 129850rs1002 C G 0.03606 0.0034 0.0115 0.7659 129799rs1003 A C 0.5128 0.045 0.038 0.2319 129830
每一列按顺序分别是SNP、编码等位基因、其他等位基因、效应等位基因的频率(freq)、效应大小(b)、标准误差、p 值、样本量
xqtl文件格式,bsed格式(类似plink软件的输出需要使用smr生成)
下面以常见的eqtl分析软件 Matrix eQTL 的输出为例
SNP gene beta t-stat p-value FDRrs13258200 ENSG00000071894.10 -1.00783189089702 -16.641554315712 2.3556801409867e-24 1.12905157909337e-18rs6599528 ENSG00000071894.10 -1.06253739134798 -15.8412867110849 2.73027622294589e-23 5.51886367106636e-18rs2272666 ENSG00000071894.10 -1.04810713295621 -15.6736668186937 4.6058755123246e-23 5.51886367106636e-18
每一列是啥就不介绍了,如果不是这个格式比如没有t-test,可以手动用beta值算,有了这个数据以后使用smr生成bsed格式的文件,需要3步
smr --eqtl-summary fastqtlnomi.txt --fastqtl-nominal-format --make-besd --out mybesd smr --beqtl-summary my_beqtl --update-esi mybigpool.esi smr --beqtl-summary my_beqtl --update-epi mybigpool.epi
其中esi是等位基因及频率信息,epi是位置和方向,了解一下就可以,关键的besd文件是二进制的,存储了beta值和se,然后就可以执行上面的smr分析了,得到我们关键的输出文件
smr.smr
ProbeID Probe_Chr Gene Probe_bp SNP SNP_Chr SNP_bp A1 A2 Freq b_GWAS se_GWAS p_GWAS b_eQTL se_eQTL p_eQTL b_SMR se_SMR p_SMR p_HEIDI nsnp_HEIDIprb01 1 Gene1 1001 rs01 1 1011 C T 0.95 -0.024 0.0063 1.4e-04 0.36 0.048 6.4e-14 -0.0668 0.0197 6.8e-04 NA NAprb02 1 Gene2 2001 rs02 1 2011 G C 0.0747 0.0034 0.0062 5.8e-01 0.62 0.0396 2e-55 0.0055 0.01 5.8e-01 4.17e-01 28
行数非常多,每一列分别是
探针 ID、探针染色体、基因名称、探针位置、SNP 名称、SNP 染色体、SNP 位置、效应等位基因、其他等位基因、效应等位基因的频率、GWAS 的效应大小、GWAS 的 SE、GWAS 的 p 值、eQTL 研究的效应大小、eQTL 研究的 SE、eQTL 研究的 p 值、 eQTL 研究的 p 值、 SMR 的效应大小、SMR 的 SE、SMR 的 p 值、HEIDI测试的 p 值以及 HEIDI 测试中使用的 SNP 数量
初学者完全不知道看什么,实际上就看2个指标,
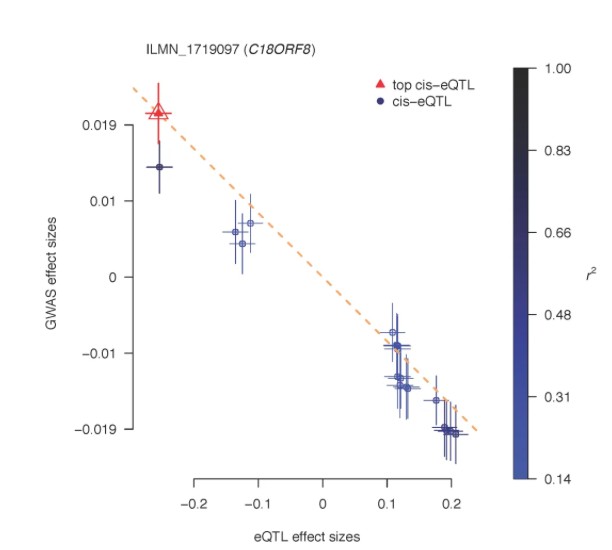
SMR检验的p值(p_SMR),这个是基因表达与表型存在因果关系的证据,一般阈值p_SMR < 8.4e-6,说明很显著
HEIDI异质性检验p值(p_HEIDI),这个是排除多效性干扰,确保因果关系是直接的,需要注意,这个和常规的p值的原假设备择假设是不同的,他的原假设是无异质性,所以需要原假设成立才行,也就是p>0.05
如果都成立,或者smr_p稍微差一点也可以接受,那么最后在看这个b_eqtl和b_gwas能否做到逻辑自洽,也就是这个等位基因的改变,对表达量的改变和表型的改变都是一致的
- 发表于 2026-01-05 17:41
- 阅读 ( 1647 )
- 分类:软件工具
