Treemix 软件:群体基因交流分析的得力工具
在群体遗传学研究中,我们常常渴望揭开不同群体之间复杂的遗传关系以及基因流动的神秘面纱。Treemix 是一款专注于推断群体间基因流的软件。它通过构建群体遗传模型,能够有效地分析群体间的遗传关系以及基因流事件,帮助我们理解物种的进化历程和群体的历史动态。今天,就让我们一起来深入了解一下这款软件。
Treemix 原理
Treemix的基本原理可以分为三个要点:
1.计算协方差:使用基因频率数据计算每对群体之间的实际协方差。
2.构建最大似然树:利用基因型频率数据构建最大似然树,并计算协方差的估计值。
3.判断基因流:通过比较实际值与估计值之间的差异,判断两个种群之间是否发生基因流。如果实际值小于估计值,则说明种群之间有基因交流,因为基因流会减少种群之间的差异
Treemix 输入文件
Treemix 需要特定格式的输入文件来开展分析。最常见的输入数据是群体等位基因频率文件。通常,这个文件包含了各个群体中不同位点的等位基因频率信息。文件格式一般为文本格式,每一行代表一个位点,每一列对应一个群体。例如:

数字分别表示不同群体在这些位点上的基因型数目。此外,还需要提供一个群体列表文件,明确每个群体的名称,确保与等位基因频率文件中的列顺序一一对应。
Treemix 的使用
1. 输入文件获得
进行群体分析的时候我们通常会得到vcf文件,在这里以vcf文件为原始数据进行输入文件的整理。
# 1.根据LD过滤,这一步会得到下面的ld.prune.in
plink --vcf treemix.vcf.gz --indep-pairwise 50 10 0.2 --out ld --allow-extra-chr --set-missing-var-ids @:# --keep-allele-order
## 根据ld.prune.in提取过滤后的位点,并且生成freq文件。
## 此处还需要treemix.cluster文件,这个文件有三列,第一列和第二列为ID名称,第三列为分组信息
plink --vcf treemix.vcf.gz --extract ld.prune.in --freq gz --missing --within treemix.cluster --out input --allow-extra-chr --set-missing-var-ids @:# --keep-allele-order
# 2. 将输出文件转换为Treemix需求的格式
## 输入文件是上一步得到的input.frq.strat.gz
## 用到的是treemix自己提供的脚本plink2treemix.py
python plink2treemix.py input.frq.strat.gz input.treemix.frq.gz
# 3.pop.order.txt
awk 'NR>1 {print}' treemix.cluster | cut -f3 | sort -u > pop.order.txt
2. treemix软件使用
treemix 参数如下:
-k [int] 如果没有做ld过滤需要指定多少个SNP为一个block
-global. Do a round of global rearrangements after adding all populations
-tf [file of nwk] Read the tree topology from a file
-m [int] 基因流次数
-root [string]
-se 权重标准误
-bootstrap
-noss 关闭亚群个体的矫正,当你的亚群中仅存在一个个体的时候可以使用
# 假设各群体间没有基因流
treemix -i input.treemix.frq.gz -m 0 -o treemix.0 -bootstrap -noss > treemix_0_log
# 假设各群体间有一次基因流
treemix -i ../input.treemix.frq.gz -m 1 -o treemix.1 -bootstrap -noss > treemix_1
输出文件
.cov.gz 文件:这是群体间等位基因频率的协方差矩阵文件,反映了群体间的遗传关系,数值大小体现关系紧密程度。
.modelcov.gz 文件:基于构建模型计算的协方差矩阵,用于评估模型对实际数据的拟合程度。

.llik 文件:记录了不同模型的对数似然值,数值越大表示模型对数据的拟合越好,可用于模型选择。

.treeout 文件:拟合树模型(fitted tree model)和迁移事件(migration events)。第一行是Newick格式的ML树,剩下行包含迁移边界(migration edges)。

.vertices.gz和.edges.gz: 包含推断图的内部结构。
.covse.gz: 协方差矩阵的标准误差(standard errors)。

结果绘图
1.树图
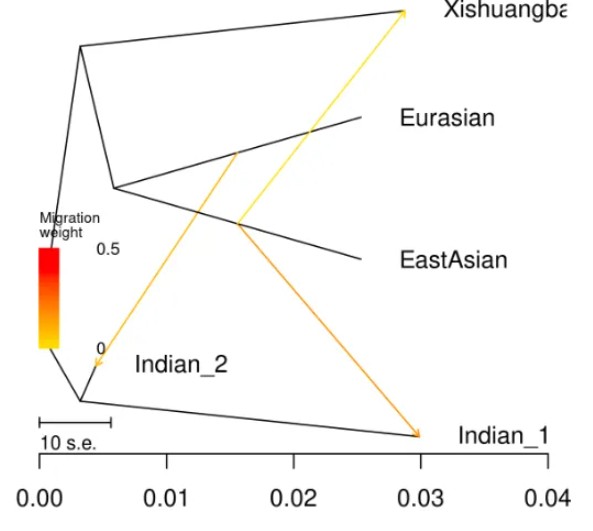
2.残差图

参考资料:
软件下载:https://bitbucket.org/nygcresearch/treemix/downloads/
https://yanzhongsino.github.io/2022/03/20/bioinfo_geneflow_TreeMix/
https://www.jianshu.com/p/92694c46cf5c
- 发表于 2025-04-30 10:31
- 阅读 ( 6257 )
- 分类:软件工具
