转录组扫盲系列--reads如何比对到参考基因组上?
上一期给大家介绍了转录组建库原理:转录组扫盲系列--转录组如何建库?建完库下一步就是测序,测序原理讲起来其实很复杂,但是我觉得测序过程少了解一点对于理解转录组数据影响不是很大,下面有个视频,大家可以了解一下小小的测序芯片方寸之地的波澜壮阔的工程之美:illumina测序原理-组学生物翻译(点此链接观看)。
数据下机后就是数据分析,在整个分析流程中我觉得最重要的步骤就是reads比对参考基因组,了解比对过程对于了解转录组的大有裨益!今天我就给大家介绍下reads是如何比对到参考基因组的。
什么是插入片段?reads?
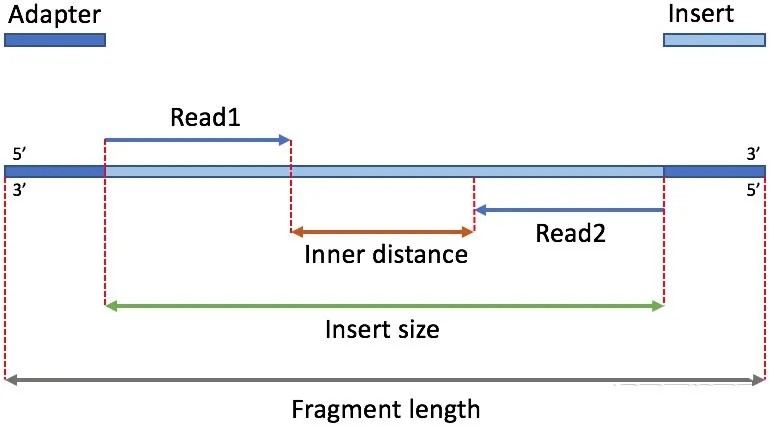
前面建库原理部分我们讲过受制于测序技术的原因,基因需要先随机打断成300bp左右的小片段然后再反转录,这些反转录好的300bp左右的cDNA称为插入片段(Insert Fragment),如下图:两端深蓝色部分是测序接头(adapter),中间淡蓝色是要测序的插入片段,插入片段长度(Insert size)指的就是这个浅蓝色部分的长度。
目前的高通量测序仪是双端测序,也就是分别从插入片段两端进行测序,每一端读取的ATCG序列称为一条reads,每条插入片段都会产生2条reads,即reads1和reads2,一个样品对应的reads1和reads2数据是分为2个压缩包存放的,我们也把这些未过滤的reads称为原始数据(raw data),过滤掉接头及低质量的reads后的数据称为clean data。
有参转录组reads比对参考基因组
参考基因组是指该物种已经破译的全基因组序列信息及注释文件,reads比对到参考基因组是数据分析的第一步,也是最重要的一步,其他所有分析内容都是基于reads比对结果分析的,如下图:
那么,reads比对是你所想的那样,根据序列互补直接比到参考基因上吗?事实上,reads比对没那么简单,应为真核生物的基因组结构复杂,外显子、内含子交替排列,并不是所有的reads只会比对到一个结构上,甚至有很多reads是横跨内含子的,这样的话怎么能比对上去呢!
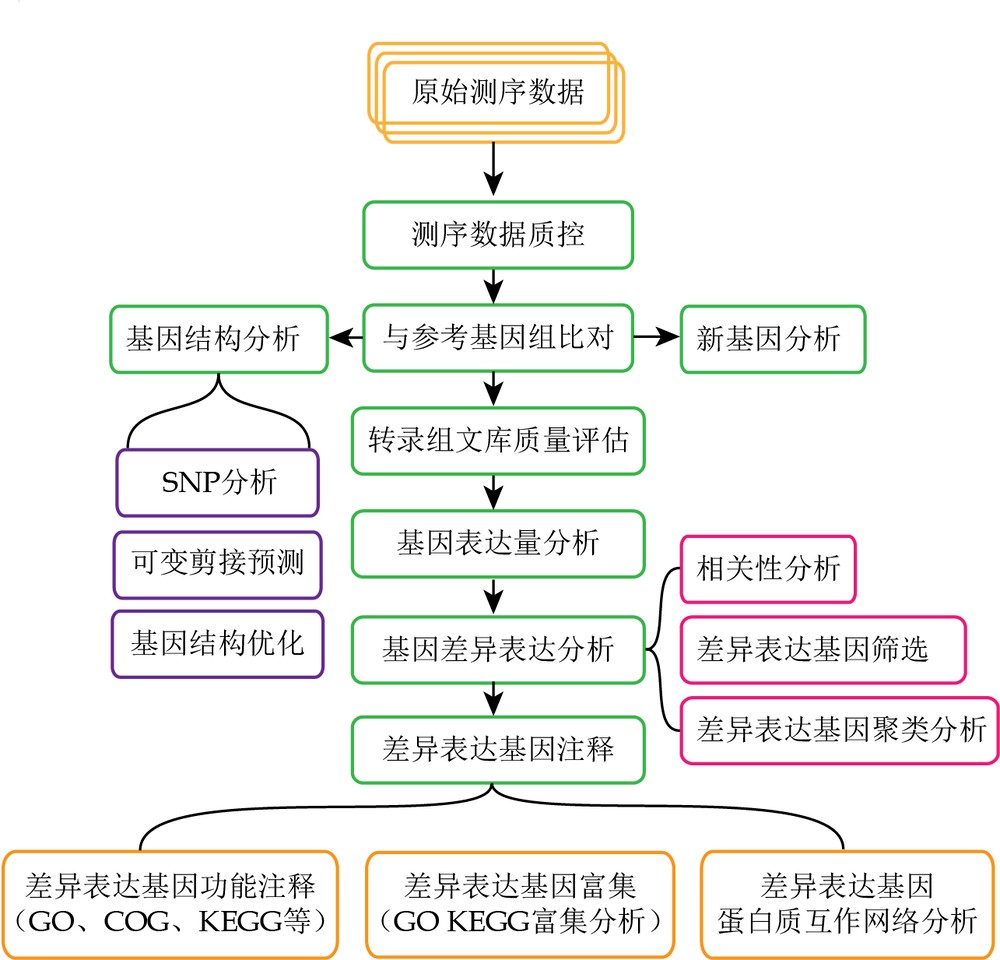
那么,现实转录组项目里是如何比对的呢?以软件Tophat2为例,reads比对有3个过程:
1. reads比对到转录组
假如参考基因组注释信息是完整的,Tophat2就会先将reads比对到该参考基因组提取出的转录本序列上,这就大大提高了比对的准确性,还可以避免序列比对到假基因上。
2. reads比对到基因组
上一步没有完全比对到转录组的read会进一步通过Bowtie2软件比对到基因组序列,在这一步比对中,只有能够连续比对到单一外显子的reads将会映射到基因组上,而比对到多个外显子的read则不会映射到基因组上。
3. 剪接比对
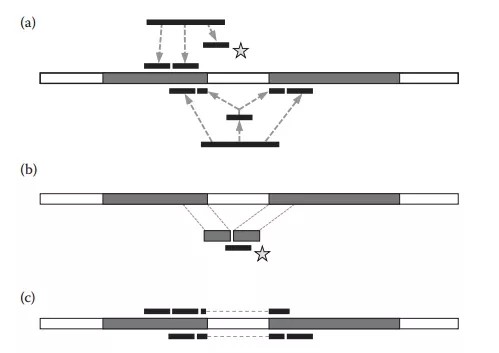
经上述2步未被映射上的read将会被片段化,通常默认大小为25bp,再次比对到基因组上,该步骤较为复杂,可总结为以下3个步骤,如下图,(a)寻找剪接位点:如果TopHat2发现reads片段化后的左右两个片段位于用户定义的最大内含子区长度范围内,TopHat2就会将read映射到整个基因组的区域以便于去寻找那些含有剪接信号(GT-AG, GC-AG, or AT-AC))的剪接位点;(b)串联间隔序列:将步骤(a)中找到的潜在的剪接位点的侧翼基因组序列被串联起来,并构建索引,未映射的read片段用Bowtie2比再次比对串联的侧翼序列。(c)重新比对:被分割的片段重新连接形成完整的reads成整个reads的跨内含子比对。
需要注意的是在步骤2 reads比对到基因组中会有一些reads会比对到内含子区域,此类reads将会用新的剪接位点的信息进行重新再比对一次。
无参转录组reads比对
无参物种没有可用的参考基因组,是如何进行reads比对的呢,如下图:我们可以看到与有参不同的是无参转录组需要先将reads拼接、组装成转录本,目前常用的组装软件是Trinity,该软件是由耶路撒冷希伯来大学和Broad研究所共同开发的一种针对无参考基因组RNA-Seq数据构建转录本的工具。
对于某个基因Trinity可能会拼接出多个对应的转录本,一般会选取其中长度最长的转录本作为Unigene来代表该基因的转录本,把所有的unigene组成的序列集当做该物种的参考基因组,然后就可以进行reads比对了。
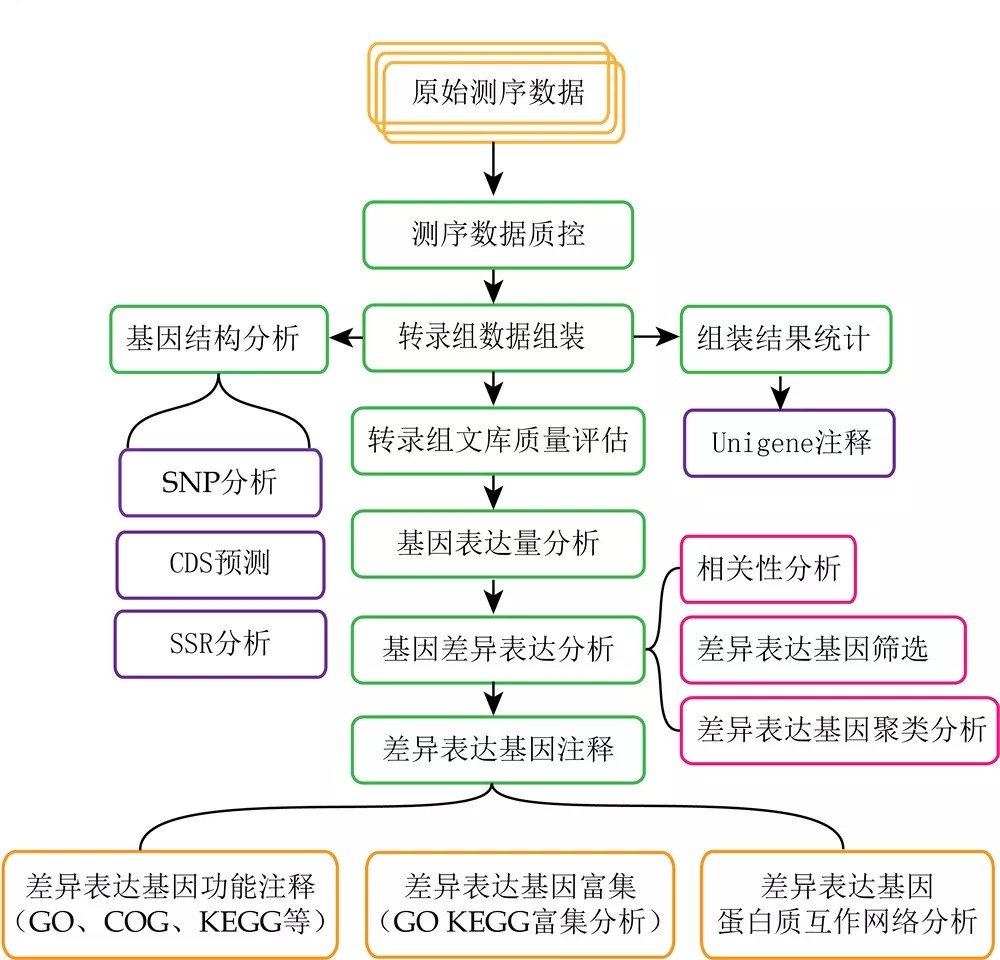
reads比对中的几个关键概念
Mapped reads:比对到参考基因组(无参物种是unigenes,下同)的reads,mapped reads并非严格要求100%比对,比对软件一般都会有一定的容错率,一般reads与参考基因组允许最大错配为2个碱基。
Multiple mapped reads:比对到参考基因组多处位置的Reads数目。
Uniq Mapped reads:比对到参考基因组唯一位置的reads。
mapping rate:比对到基因组的reads占clean reads的比值;比对率会随着亲缘关系、基因组组装质量、测序质量、有无污染等有所波动,一般mapping rate大于60%,再低的话就要考虑进行无参组装了。
BAM文件:reads比对到参考基因组后,会得到一个以sam或bam为扩展名的文件,而bam就是sam的二进制文件,也就是压缩格式的sam文件,里面存储了reads的比对信息,更多信息请参见:你了解SAM和BAM文件吗。
好了,reads比对今天就介绍到这里,希望对您的学习有所帮助!下一期我会继续给大家分享如何利用reads计算基因的表达量,感兴趣的朋友请持续关注!
日常科研中你我经会常遇到看不懂的图表,不会挖掘的数据,没有思路的文章,沟通不畅的个性化分析,求人不如求己,一切痛点都能解决:
1. 单细胞/空间转录组正在大火,高分文章必备,0基础学单细胞/空间转录组分析,做更牛的个性化分析,绘更漂亮的图,点这个链接:单细胞/空间转录组分析实操
2. 2区文章发文新思路,0实验就能发文章,省钱省时间,套路化内容好写易发表,0基础学习最新版基因家族分析/泛基因家族分析链接:最新基因家族分析 ;最新版泛基因家族分析
3. 转录组越做越普遍,实验必备,看不懂结果?不会深入分析?自学都可以搞定,学习链接:有参转录组自主分析实操 ;转录组与代谢组结果解读/个性化数据分析 ;
4. 代谢组分析硬件要求不高,个人电脑就可以分析,0基础学习链接:代谢组分析实操
3. 现在才是发表T2T基因组/泛基因组文章的好时机,成本低,好发表,做好高级个性化分析发表高分1区不是梦!T2T基因组/比较基因组/泛基因组分析学习链接:T2T基因组组装与注释分析;动植物泛基因组分析 ;比较基因组分析
4. 群体重测序遗传进化分析+GWAS文章,篇篇10+分,缩短分析周期,提升文章亮点,勇敢冲击NGS顶刊,学习链接:群体遗传进化GWAS分析 ;
5. 传统图位克隆研究方法经久不衰,有遗传群体,有性状,自己就可以做,学习链接:遗传图谱构建与QTL分析
6. 微生物16S/18S/ITS多样性分析和宏基因组分析,学习链接:宏基因组分析 ;微生物16S/18s/ITS多样性分析
7. 细胞器基因组与比较基因组分析是真正的无需实验就可以发表2区期刊的文章思路,成本低,文章好发,性价比极高,学习链接:线粒体基因组分析实操 ;叶绿体基因组分析实操
8. 数据分析硬件利器,个人分析能力的倍增器,好用便宜的云生信服务器,专门为高通量测序数据分析而生,了解链接:云生信服务器
9. 免费好课,个人能力提升,生信入门,不可错过,内容涵盖:生信语言与工具基础、R语言绘图、常见科研绘图、数据处理技能等,免费好课总入口:
10. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击进入组学大讲堂网校学习
- 发表于 2021-04-26 19:13
- 阅读 ( 34769 )
- 分类:转录组
