16S聚类还是降噪?
前言:
利用聚类 (Clustering) 得到OTU的方法近些年已经广泛应用于扩增子高通量测序数据的分析过程中。经典的Uparse就是通过序列之间的相似度为阈值(一般选择97%)将序列划分为OTUs。利用Semantic Scholar进行查询,Uparse目前总引用次数为6301,2015-2017年平均引用次数2125次。
一. DADA
DADA全称Divisive Amplicon Denoising Algorithm,旨在通过降噪得到不含扩增与测序错误、不含嵌合体的生物学序列。DADA使用454测序数据进行了测试,结果表明可得到更少的假阳性。2016年在DADA基础上推出的DADA2可用于分析Illumina平台数据。其分辨率达到单核苷酸精度[2]。
DADA2算法是基于Illumina扩增序列的错误的模型。其核心算法是:所有不同的序列都作为一个partition,先指定丰度最高的序列为中心。所有的partition都和中心序列进行比较,计算丰度的p-value。p-value定量了一条序列被判定为扩增错误的可能性。如果最小的p-value低于设定的阈值,就会得到一个新的partition。所有的序列再与新的partition比较,重新计算P-value。如此不断进行迭代,直到每条序列都被划分为最可能的partition中。
二. Unoise
针对DADA2的文章指出自己的精确度比Uparse好,Edgar,R.C.大神马上推出了进的降噪方法Unoise2,旨在提升Illumina平台16S与ITS扩增子测序的错误校正能力,并在文章中证明了Unoise2的准确度要优于DADA2 [3]。
Unoise2只针对Illumina产生的数据,不支持处理454, Ion Torrent 或者 PacBio 产生的数据。Unosie2主要有两个作用:去除嵌合体;去除测序和PCR过程的点错误。
Unoise2算法简介:
序列按照丰度多少进行降序排列,最高丰度的序列先定义为中心序列(centroid)。假设C是一个cluster的中心序列,丰度为aC。M是这个cluster中的一条序列,丰度为aM。d是Levenshtein distance,表示M与C之间由于碱基的替换与gap综合起来的距离 (图2)。定义skew(M,C)=aM/aC。若d和skew(M, C)足够小,则可认为M是cluster C中伴有d个点错误的不正确读长。
Unoise2默认保留丰度在4以上的序列(Unoise3中为8),低丰度的序列由于包含错误的可能性更高而被丢弃。
安装地址:(32位免费)
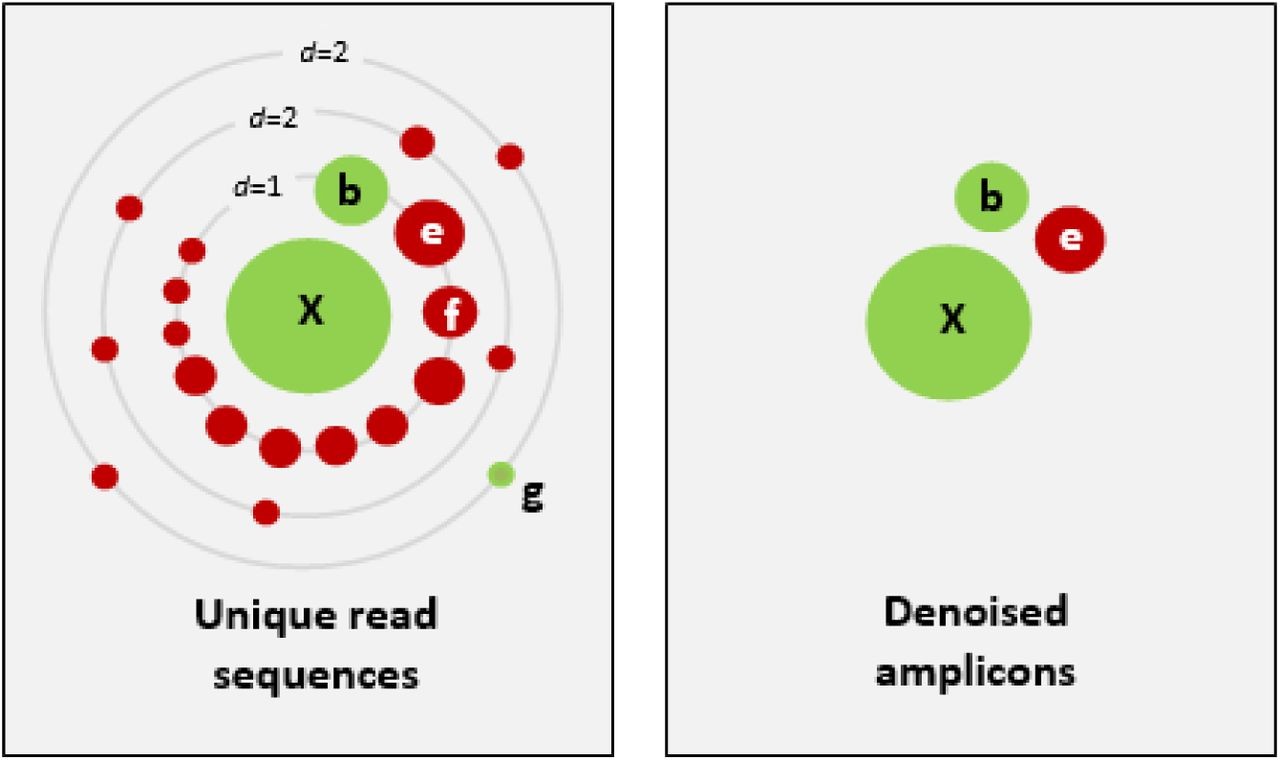
X为一条高丰度序列,周围存在很多低丰度序列。d为序列的差异程度。周围的点为不同的序列,点的大小代表其丰度。绿色的点为正确的生物学序列,红的点代表含有一个至多个错误点的序列。右图表示降噪后的序列。E代表高丰度的错误序列,被错误的保留下来,f表示错误序列被准确的丢弃,g表示丰度较低的正确序列被错误的删除。b,e,f丰度相近而降噪结果不同,表明了降噪过程普遍存在的一个问题:如何设定阈值,准确的区分正确与错误序列。
Unoise目前已经升级到Unoise3版本整合在Usearch10中,使用起来相当方便:
usearch -unoise3 uniques.fa-zotus zotus.fa -tabbedout unoise3.txt -minsize 8
到底应该用Uparse还是Unoise3? Edgar,R.C.给出了他的建议 [4]:
Uparse算法采用97%相似度聚类得到OTUs,Unoise算法通过降噪得到zOTUs。
a. Uparse得到的是所有正确的生物学序列的子集,每两条序列之间的相似程度在97%以上(对应USEARCH中的cluster_otuscommand)。
b. Unoise得到的是所有的正确生物学序列(对应USEARCH中的unoise3 command)。
当然每一种算法都不是完美的,因此总是存在一些错误。如Uparse会丢失一些具有意义的生物学序列,而Unoise在降噪过程中由于种内差异,对结果亦会造成影响,可能得到2个或多个OTU。但是如果要检测菌株不同的表型,这反倒又成为了优势。如果只能选择一个,Edgar,R.C.推荐Unoise。目前32位的Usearch是免费的,若数据量小可直接下载32位的使用。大数据需要购买64位的Usearch10进行分析。
三. Deblur
另一种新颖的不通过clustering而得到OTU的方法是2016年发表的Deblur[5]。此方法已经被用于处理地球微生物组计划得到的全球数据,今年11月刚刚发表在Nature上 [6]。DADA2和Unoise2只能对混合样本进行操作,Deblur的优势是不仅可以对混合样本进行操作,还能对单个样本进行操作。Deblur分辨率也能够达到单核苷酸精度。
文章结果表明,Deblur得到的结果稳定性优于DADA2。另外Unoise结果受到测序批次和测序中心的影响较大,基于此文章指出不推荐Unoise2 。
运行速度上Unoise2最快,Deblur低一个数量级,DADA2再低一个数量级 。
算法简介:
1.序列丰度从高到底排序;
2.基于Hamming distances预测的错误读长被删除。碱基的插入缺失最大概率设定为0.01,平均读长错误率为0.5%;
3.不断删除错误读长,当一条序列丰度降为0,整条序列被删除;
总结
不同的方法虽然结果存在差异,但是核心的降噪思想是一致的。具体选择哪种方法还需要研究者谨慎考虑。我刚进实验室的时候还是Uclust和Uparse两强相争的时代。但是改变总是好事,新的方法层出不穷,让我们不断地认识新概念、发现新问题、挖掘新信息,更好的探索微生物的神奇世界。
课程推荐:微生物扩增子分析课程实操 微生物16S/ITS/18S分析原理及结果解读
一个小问题
不同方法对丰度阈值的选择存在差异。DADA2和Uparse都推荐去掉singletons。Deblur默认去掉singletons。Unoise2默认去掉丰度小于4的序列,Unoise3默认为8。不同方法的阈值存在差异,而这一问题目前好像还没有统一的结论。去掉低丰度的序列会去掉一些错误,但同时一些稀有物种也会被丢弃。这就需要依据研究目的在准确性和灵敏性上进行权衡,合理的选择阈值。
参考文献:
[1] Edgar, R.C. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. (2017). doi:https://doi.org/10.1101/192211
[2] Callahan,B. J., P. J. McMurdie, M. J. Rosen, A. W. Han, A. J. A. Johnson and S. P.Holmes (2016). "DADA2: High-resolution sample inference from Illuminaamplicon data." Nature Methods 13(7):581-+.
[3] Edgar,R.C.(2016) UNOISE2: improved error-correction for Illumina 16S and ITS ampliconsequencing.doi: http://dx.doi.org/10.1101/081257
[4] http://www.drive5.com/usearch/manual/faq_uparse_or_unoise.html
[5] Amir,A., D. McDonald, J. A. Navas-Molina, E. Kopylova, J. T. Morton, Z. Z. Xu, E. P.Kightley, L. R. Thompson, E. R. Hyde, A. Gonzalez and R. Knight (2017)."Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns."Msystems 2(2).
[6] Thompson LR, Sanders JG, McDonald D,Amir A, Ladau J,Locey KJ et al (2017). A communal cataloguereveals Earth’s multiscalemicrobialdiversity. Nature.
中国科学院生态环境研究中心
环境生物技术重点实验室
邓晔 研究员课题组发布
- 发表于 2020-11-02 20:59
- 阅读 ( 7260 )
- 分类:宏基因组
