运行代码
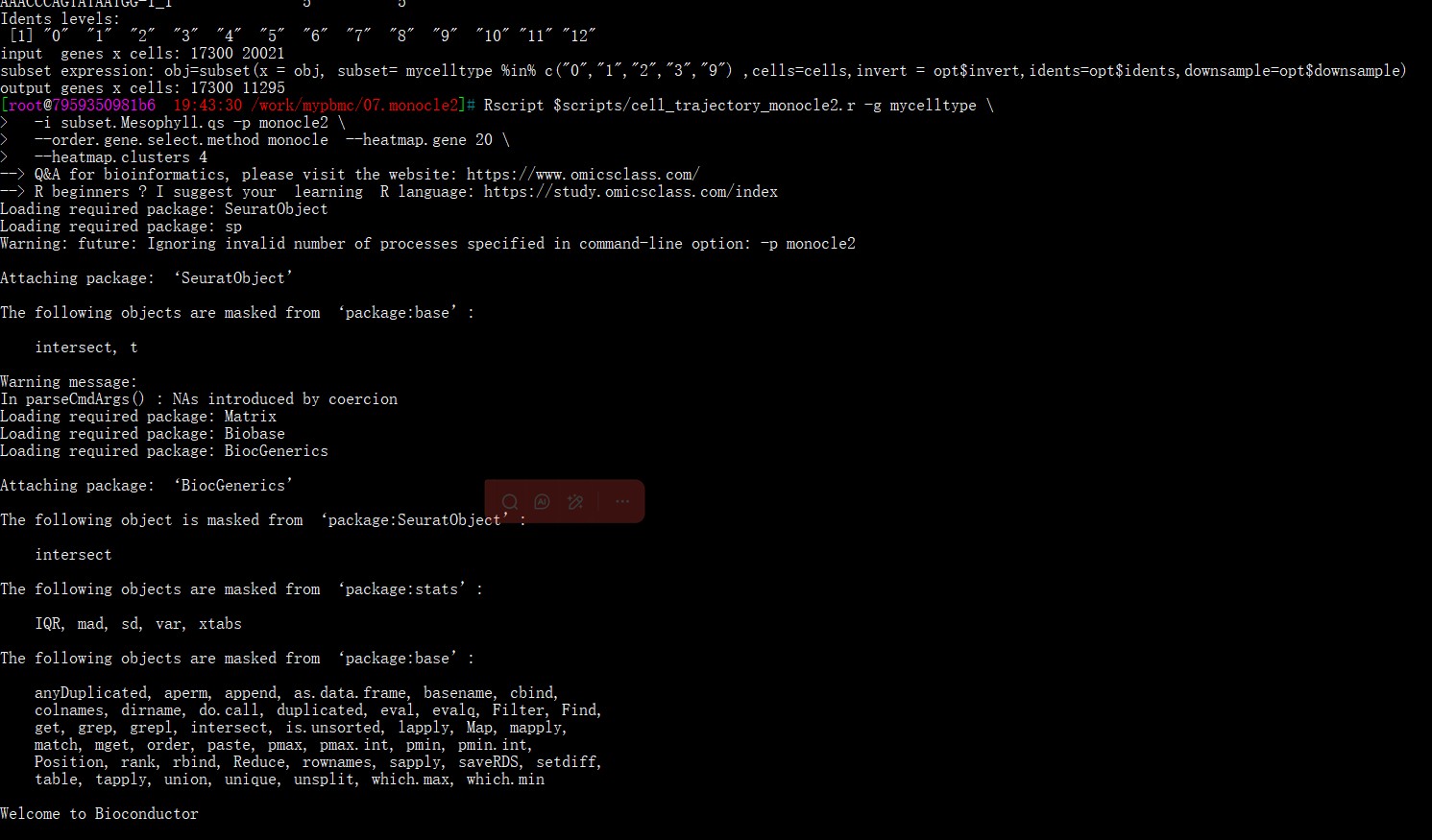
Rscript $scripts/cell_trajectory_monocle2.r -g mycelltype \
-i subset.Mesophyll.qs -p monocle2 \
--order.gene.select.method monocle --heatmap.gene 20 \
--heatmap.clusters 4
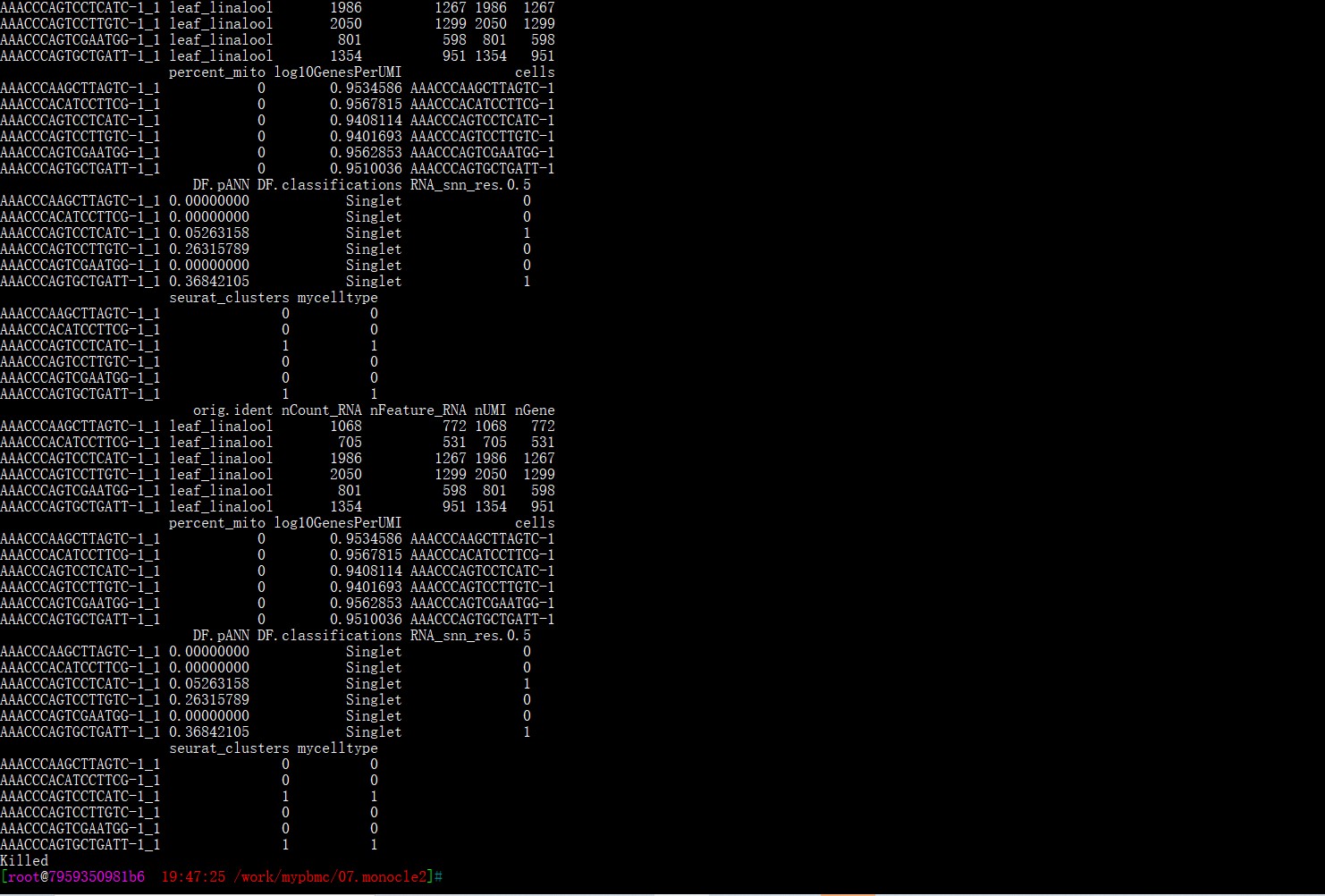
出现killed,代码运行不了
电脑内存不够就会被KILL,查看内存使用情况: free -h
你看看这个设置docker内存使用:https://www.omicsclass.com/article/1413
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!