fastp也有同样的问题说明 输入的fastq数据有问题,不完整或者格式不对你再检查一下;
转录组上游分析之定量比对Hisat2出现内部异常错误
转录组上游分析(Linux)之定量比对Hisat2出现内部异常错误,不知道哪里错了?
如图下:
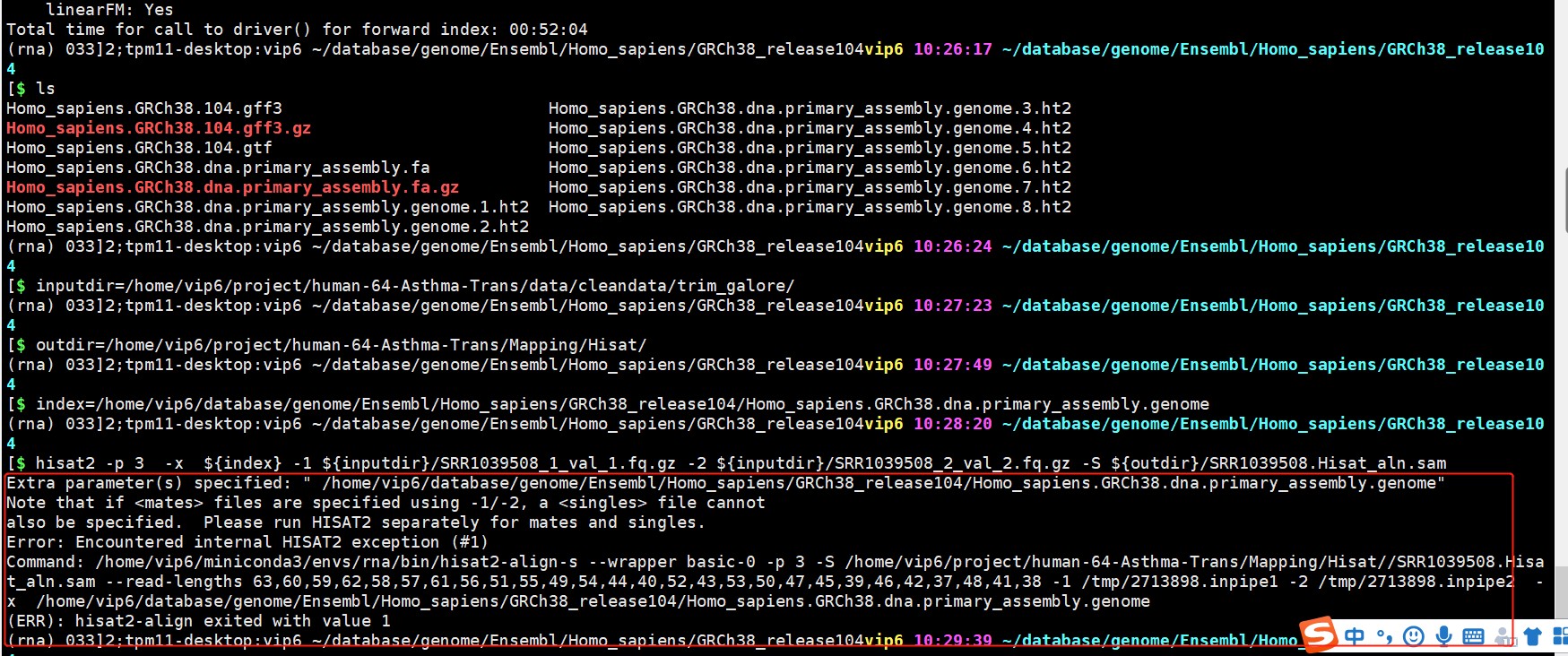
下面为上图的红圈error信息:
Extra parameter(s) specified: "?/home/vip6/database/genome/Ensembl/Homo_sapiens/GRCh38_release104/Homo_sapiens.GRCh38_release104.genome"
Note that if <mates> files are specified using -1/-2, a <singles> file cannot
also be specified. Please run HISAT2 separately for mates and singles.
Error: Encountered internal HISAT2 exception (#1)
Command: /home/vip6/miniconda3/envs/rna/bin/hisat2-align-s --wrapper basic-0 -p 3 -S /home/vip6/project/human-64-Asthma-Trans/Mapping/Hisat//SRR1039508.Hisat_aln.sam --read-lengths 63,60,59,62,58,57,61,56,51,55,49,54,44,40,52,43,53,50,47,45,39,46,42,37,48,41,38 -1 /tmp/2597247.inpipe1 -2 /tmp/2597247.inpipe2 ?-x ?/home/vip6/database/genome/Ensembl/Homo_sapiens/GRCh38_release104/Homo_sapiens.GRCh38_release104.genome
(ERR): hisat2-align exited with value 1
数据用的是trim_galore进行数据过滤的,Hisat2已经构建好索引,1/2指定路径准确,fq.gz文件完整,改SRR1039508根据SRA数据库得知为双端测序,但是一直是这样的错误,不知道怎么回事
另外,我用了fastp进行数据过滤,也得到同样的bug,所以不是数据过滤的问题
同时我把-1和-2的$变量目录改成绝对目录,也是不行,还是同样bug
难道是索引问题?还是Hisat2版本识别不了的问题?
请大家帮忙看一下,是哪里出错了,谢谢大家!
