物种基因组注释不好的话,有可能比对率较低。再有检查一下建立的参考基因组索引是否正确,基因数量是否全。
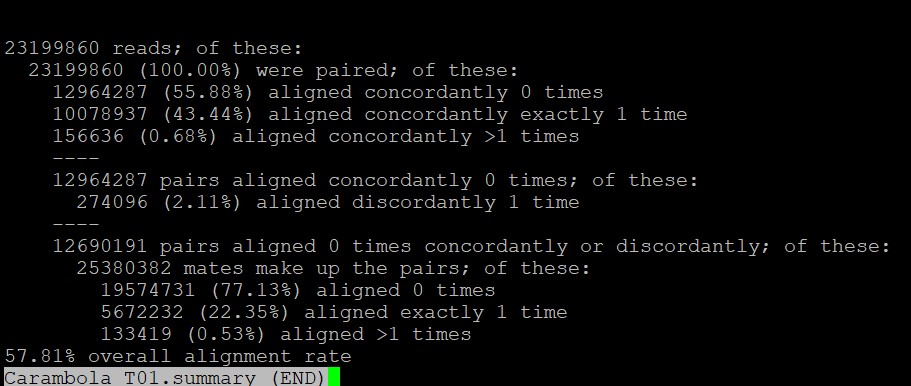
老师,你好。在做有参转录组分析,准备的文件有基因组注释文件gtf,cds,pep(其中cds,pep都是fasta格式的序列文件),命令如下。其中第一个截图是参考pep文件,比对率为0,;第二个结果是参考cds文件,比对率为57.81%,比较低,这是哪里出了问题吗。另外所以准备的文件应该是cds文件而不是pep文件是吗
# 对基因组构建HISAT index
echo "Step1: build reference index "
hisat2_extract_splice_sites.py /bioData/run_data/lixp/work/GZ_yangtao2/refs/Yangtao2.coding.gene.V1.0.20190227.gtf > /bioData/run_data/lixp/work/GZ_yangtao2/refs/splicesites.tsv
hisat2_extract_exons.py /bioData/run_data/lixp/work/GZ_yangtao2/refs/Yangtao2.coding.gene.V1.0.20190227.gtf > /bioData/run_data/lixp/work/GZ_yangtao2/refs/exons.tsv
hisat2-build -p 8 --ss /bioData/run_data/lixp/work/GZ_yangtao2/refs/splicesites.tsv --exon /bioData/run_data/lixp/work/GZ_yangtao2/refs/exons.tsv /bioData/run_data/lixp/work/GZ_yangtao2/refs/Yangtao2.coding.gene.V1.0.20190227.fa /bioData/run_data/lixp/work/GZ_yangtao2/refs/Yangtao2.coding.gene.V1.0.20190227
# reads 与基因组进行比对
hisat2 -p 8 --rg-id=Carambola_T01 --rg SM:Carambola --rg LB:Carambola_K007-01-T01 --rg PL:ILLUMINA --rg PU:CXX1234-ACTGAC.1 -x /bioData/run_data/lixp/work/GZ_yangtao2/refs/Yangtao2.coding.gene.V1.0.20190227 --dta --rna-strandness RF -1 /bioData/run_data/lixp/work/rna_seq_data/Carambola_K007-01-T01_good_1.fq.gz -2 /bioData/run_data/lixp/work/rna_seq_data/Carambola_K007-01-T01_good_2.fq.gz -S /bioData/run_data/lixp/work/GZ_yangtao2/2.alignment/hisat2/Carambola_T01.sam 2>/bioData/run_data/lixp/work/GZ_yangtao2/2.alignment/hisat2/Carambola_T01.summary

2 个回答

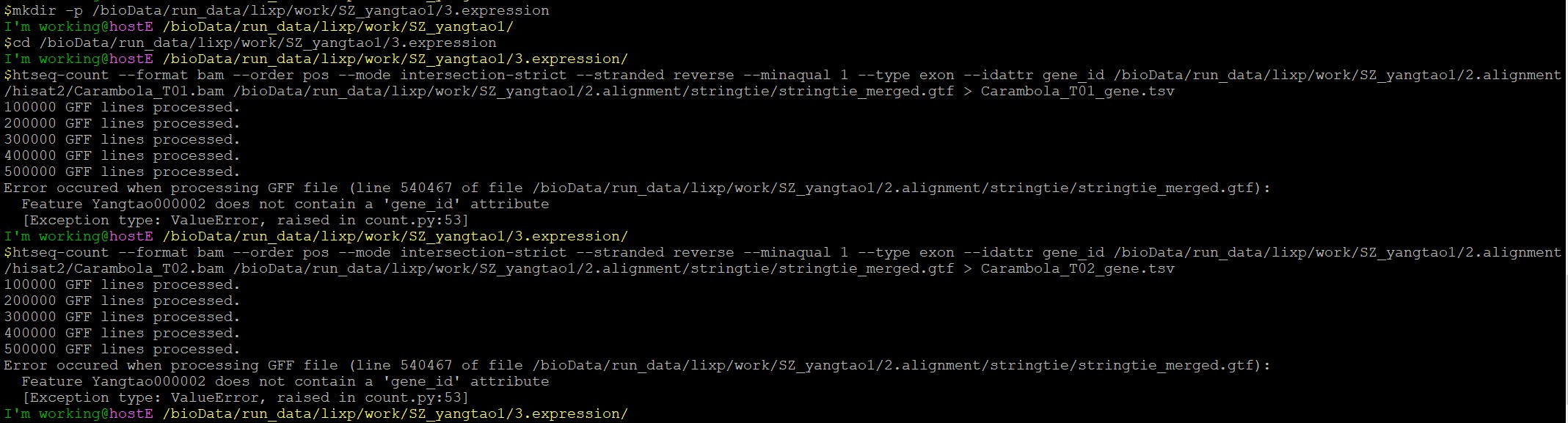
老师,这个问题是用错了文件。但是在弄基因表达定量的时候出现了这个问题,请问是什么原因呀,是格式不对吗