5 mutmap混池定位的测序要求及相应的原理?优势在哪里?
最佳答案 2018-07-30 13:05
mutmap分析原理如下图:
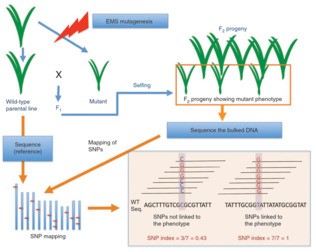
MutMap适合对EMS诱变的隐性突变基因进行分析。通过EMS诱变和自交得到纯合体后,将突变体和其亲本回交得到F1,F1自交得到的F2后代会出现表型的分离,得到野生型表型群体和突变体表型群体。对这两个群体的DNA分别进行等量混合,得到野生型DNA混池和突变体DNA混池。将两个混池分别进行DNA测序,得到野生型和突变型混池测序变异SNP信息;突变为隐性,根据遗传学定律,在F2群体中,与耐盐表型不相关的突变会随机的分布在F2群体个体中,因此,与目标性状不相干的SNP会以:野生型类型:突变体类型=1:1的比例进行分离,而导致突变体表型的SNP,受到了人为选择,在突变体混池中所有的个体都是是纯合的。
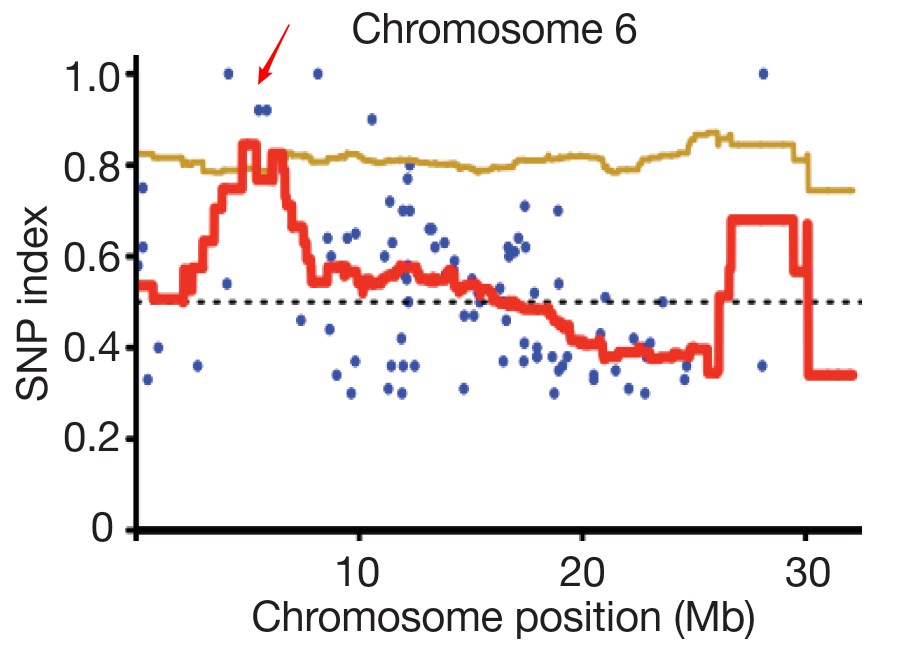
为了方便分析,作者定义了一个参数SNP-index,突变体类型的SNP所占的比例(也就是上图中右下角图示)。那么在突变位点,SNP-index为1,越往两侧,SNP-index越小,并最终接近于0.5。对SNP-index进行滑窗作图后,就会出现一个峰,该处就是连锁区域。在附近进行候选基因的筛选和排查,可以比较容易找到突变基因。
 问题:SNP位点一般为两个等位基因,与参考基因组比较我怎么知道哪个碱基是突变的?
问题:SNP位点一般为两个等位基因,与参考基因组比较我怎么知道哪个碱基是突变的?答:作者利用重测序的方法,对野生型品种(Hitomebore)进行重测序,将read比对到参考基因组日本晴(Nipponbare )上,将Hitomebore和Nipponbare 之间的差异进行替换,得到Hitomebore的参考基因组,如果我们的突变型测序结果比对到Hitomebore参考基因组上,与参考基因组不同的碱基即为突变碱基。以下是作者原文:
reference sequence was constructed by replacing nucleotides in Nipponbare with those of Hitomebore at the 124,968 SNP positions identified between the two cultivars by alignment of 12.25 Gb of Illumina Hitomebore short reads to the Nipponbare reference genome
要使用mutmap的定位方法:
1.一般为隐性突变,最好是EMS诱导的点突变;
2.必须测野生型基因组
3.必须有回交过程:纯合之后再与野生型回交,再自交得到F2.
优势:
只测野生型亲本和突变型混池就可以分析;
参考文献:
https://www.nature.com/articles/nbt.3188
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
