pacbio 三代全长转录组数据分析流程 Iso-Seq 3
pacbio 三代全长转录组数据分析流程 Iso-Seq 3
- 2
- 3
- microRNA
- 发布于 2018-08-03 10:54
- 阅读 ( 24471 )
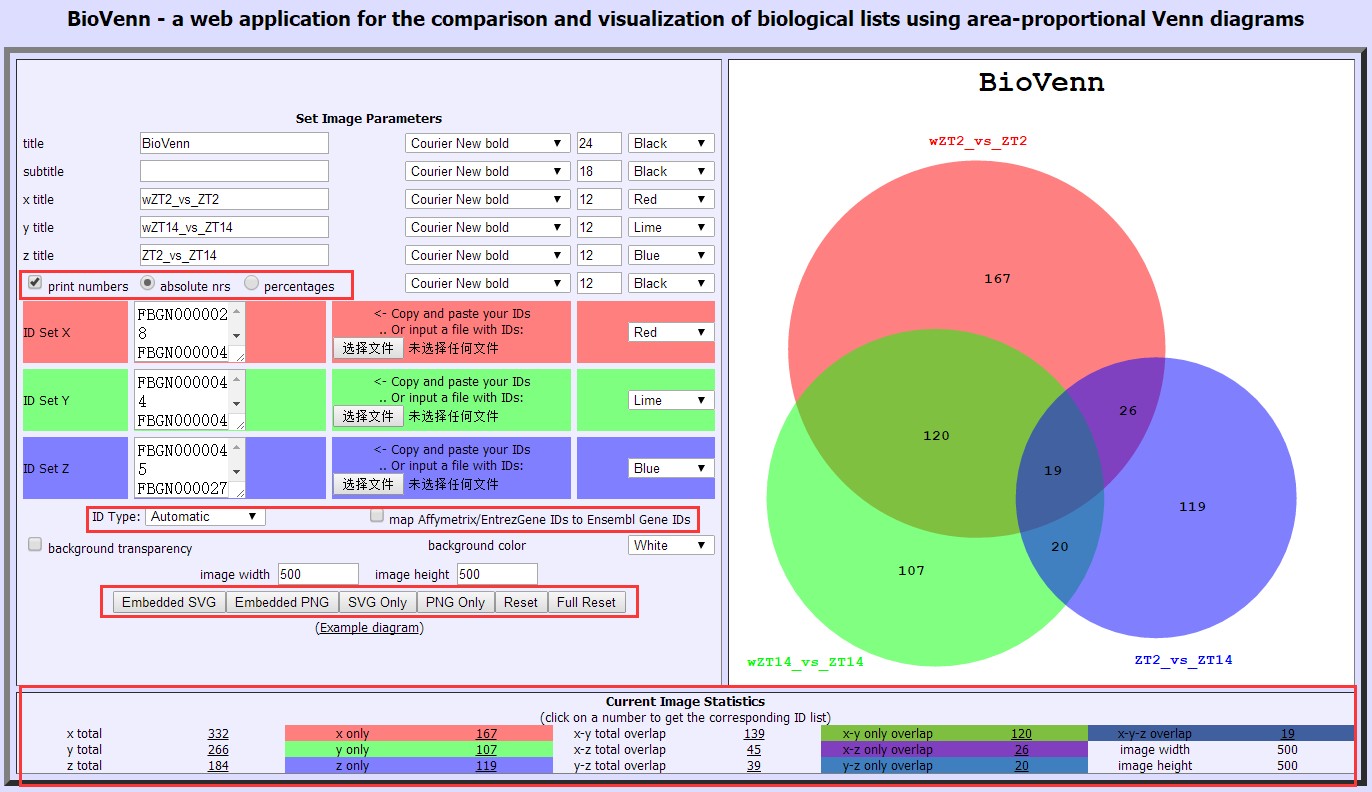
做韦恩图?自己搞定!
韦恩图又称文氏图,是科研文章中最常见的图,可以用来表示多个数据集之间的大致关系。小编来介绍几种常用在线绘制韦恩图的工具,简单易学!
- 2
- 3
- 安生水
- 发布于 2018-07-26 11:08
- 阅读 ( 33070 )
Rstudio 防止中文乱码(保存与显示)
为了防止代码文件出现中文乱码,一般在Rstudio中,都要求将文件保存为UTF-8格式,同时打开显示也要求以UTF-8格式进行显示,相应的设置方法如下: 保存文件: 进入Tools——Global Options,选...
- 0
- 3
- Daitoue
- 发布于 2018-07-17 11:57
- 阅读 ( 22667 )
转录组测序样品间的相关性有何意义?如何计算?
样品间的相关性反应了样品间的相似程度,即不同处理或组织的样品在表达水平方面的相似度。相关系数越 接近1,样品间的相似度越高,样品间的差异基因也越少。生物学重复间的样品的相关系数...
- 0
- 3
- landy
- 发布于 2018-06-28 14:02
- 阅读 ( 14787 )
MEGA构建进化树遇到报错信息
最近又有人遇到mega构建进化树时报错,所以分享一下这个经验给大家。 Oops!MEGA has encountered an error and cannot continue with this analysis:Some pairwise distances could not be estimated.For example,between sequences 23 and 11.
- 7
- 3
- omicsgene
- 发布于 2018-06-22 14:21
- 阅读 ( 40696 )
你懂荧光定量PCR吗?要点一二三帮你解惑
荧光定量虽小,细节问题挺多!正所谓“千里之行始于足下,万里之堤溃于蚁穴!”让我们从一做起!
- 3
- 3
- 生信老顽童
- 发布于 2018-06-20 10:27
- 阅读 ( 4356 )
下载TCGA基因表达量文件,应该选Count,FPKM还是FPKM-UQ ?
下载TCGA基因表达量文件,应该选Count,FPKM还是FPKM-UQ ?
- 3
- 3
- microRNA
- 发布于 2018-06-15 14:55
- 阅读 ( 15741 )
MCScanX 安装报错,支持64位系统
今天有学员问 MCScanX 安装问题,我试了下,果然有错误。 make报错: g++ struct.cc mcscan.cc read_data.cc out_utils.cc dagchainer.cc msa.cc permutation.cc -o MCScanX msa.cc: In fun...
- 0
- 3
- omicsgene
- 发布于 2018-05-16 21:20
- 阅读 ( 9589 )
pfam上不去 打开后搜索没有反应
pfam打开不了解决办法,Pfam这个主页里,这些搜索项目点击没有反应,是咋回事呢
- 0
- 3
- omicsgene
- 发布于 2018-05-09 08:33
- 阅读 ( 9053 )

如何让生信大咖解决自己的问题
在工作和学习过程中,总是会遇到不少问题需要在论坛或者群里进行提问,但是提问者和回答者之间经常会发生不愉快的事情。发生这种情况的主要原因在于提问者不知道如何准确、全面的提出问题,使得...
- 1
- 3
- omicsgene
- 发布于 2018-05-03 16:08
- 阅读 ( 7354 )
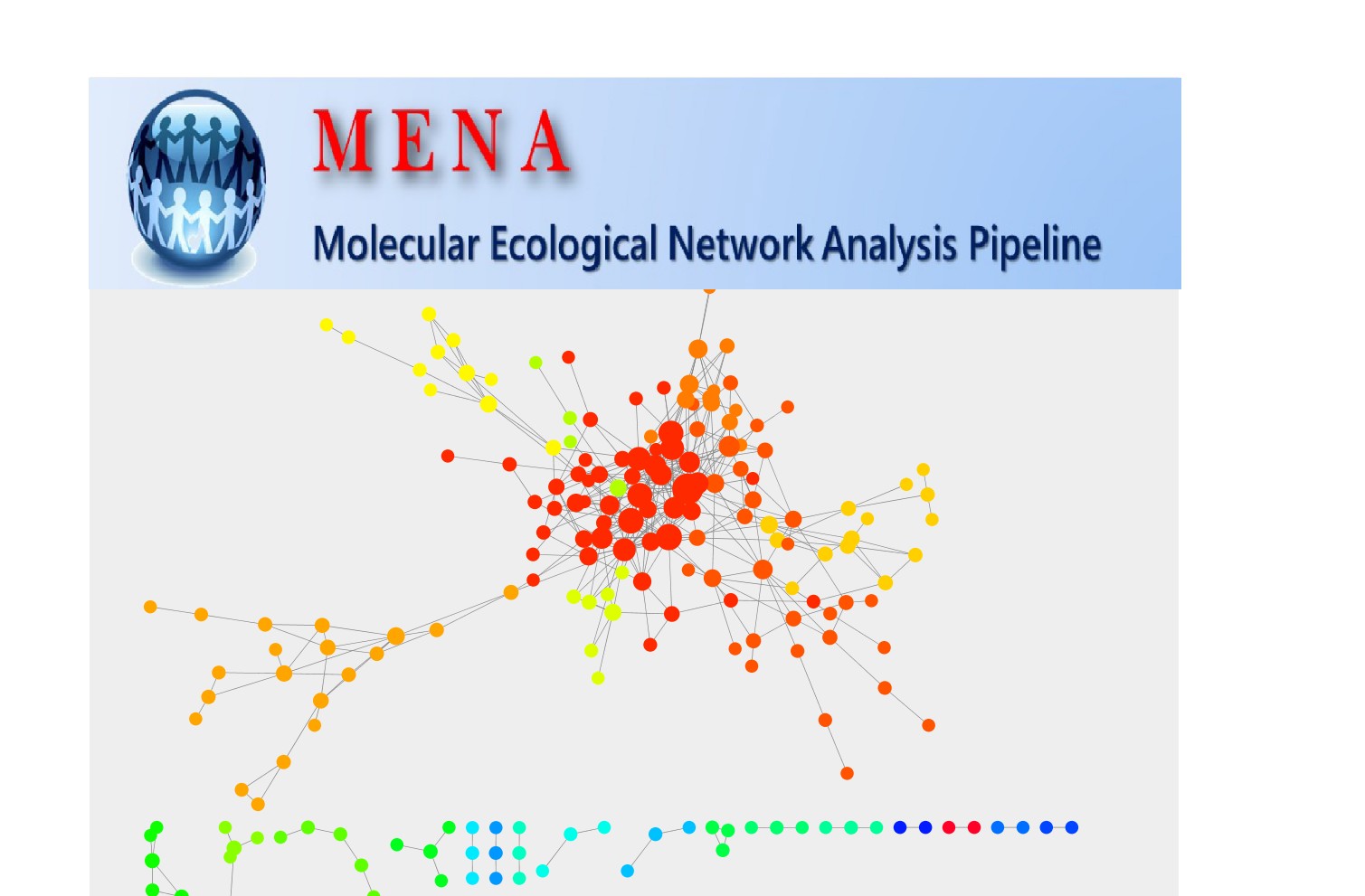
OTU互作网络分析用MENA
MENA,一个可以利用OTU数据进行在线分子生态网络分析的网站,帮你搞定OTU互作网络图。
- 1
- 3
- Daitoue
- 发布于 2018-04-22 21:19
- 阅读 ( 24036 )
两分钟看懂基因组注释GFF文件
随着大数据时代的到来,很多生物科研工作者都接触了基因组相关实验。在做数据分析的时候,会用到一个很重要的文件,就是基因组的注释文件,也就是今天分享内容的主角GFF文件! 什么是GFF文件...
- 5
- 3
- landy
- 发布于 2018-04-22 21:00
- 阅读 ( 33867 )
两分钟做一张让人羡慕的系统进化树图!
使用FigTree制作漂亮的系统发育树。这款软件需要在JAVA环境下运行,所以使用前记得要安装JAVA。 第一步:选择合适的树形 使用File – open 打开树文件(一般为.nwk文件),在左上方绿色...
- 8
- 3
- landy
- 发布于 2018-04-22 18:38
- 阅读 ( 34822 )
