cactus 软件:从基因组比到泛基因组分析
Cactus 是由 UCSC(加州大学圣克鲁兹分校) 开发的 全基因组比对工具(Whole Genome Alignment, WGA),采用 层次化比对算法(Progressive Alignment),能够高效处理跨物种或同物种的基因组比对任务。Cactus的核心是全基因组比对工具(Whole Genome Alignment),但它独特的层次化比对算法使其成为构建泛基因组(Pangenome)的基础工具。它通过比对多个基因组,识别保守区域和变异位点,从而为泛基因组分析提供输入数据。
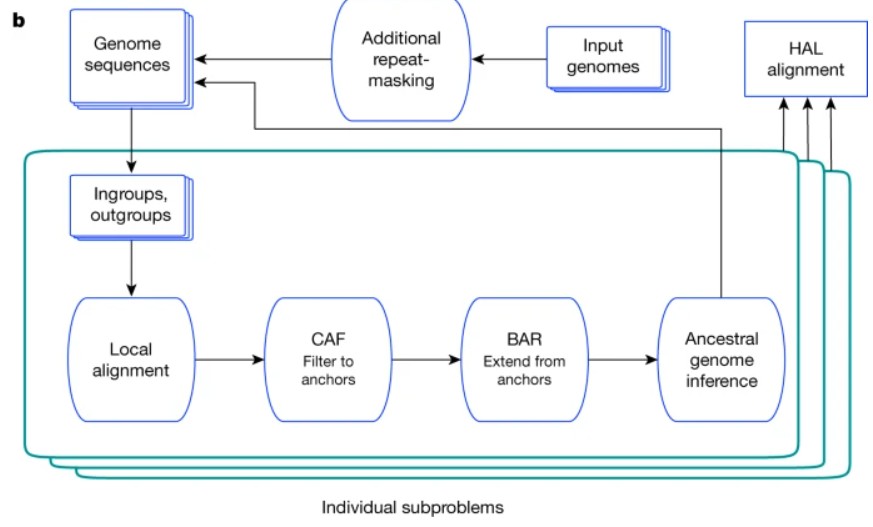
01 Cactus主要用在哪里
1 经典应用:全基因组比对
1.1 跨物种比较(如人-小鼠-鸡):分析保守区域、基因家族演化。
1.2 同物种群体分析(如不同水稻品种):检测SNP、结构变异(SV)、存在-缺失变异(PAV)。
2 延伸应用:泛基因组(Pangenome)构建
Cactus的比对结果(HAL格式)是 图泛基因组(Graph Pangenome) 的基础输入,例如:
2.1 通过工具链(如hal2vg)转换为变异图(Variation Graph)。
2.2 结合minigraph-cactus流程直接构建泛基因组。
02 下载安装方法
下载:
软件github:https://github.com/ComparativeGenomicsToolkit/cactus
选择的是Pre-compiled Binaries Linux Tarball这个版本:
wget https://github.com/ComparativeGenomicsToolkit/cactus/releases/download/v2.9.9/cactus-bin-v2.9.9.tar.gz
安装:
# 解压缩
tar -xzf cactus-bin-v2.9.9.tar.gz
# 进入文件夹
cd cactus-bin-v2.9.9
# 要构建 python virtualenv 并激活,执行以下步骤。这需要 Python 版本 >= 3.9
# 在当前文件夹下面创建了一个venv-cactus-v2.9.9的文件夹,以后激活也得记得环境所在的路径
virtualenv -p python3 venv-cactus-v2.9.9
# 把现在的环境给到虚拟环境
printf "export PATH=$(pwd)/bin:\$PATH\nexport PYTHONPATH=$(pwd)/lib:\$PYTHONPATH\nexport LD_LIBRARY_PATH=$(pwd)/lib:\$LD_LIBRARY_PATH\n" >> venv-cactus-v2.9.9/bin/activate
进行配置:
# 激活环境
source venv-cactus-v2.9.9/bin/activate
# 执行一些下载的指令
python3 -m pip install -U setuptools pip wheel
python3 -m pip install -U .
python3 -m pip install -U -r ./toil-requirement.txt
# 某些工具需要 ,并且不包括在内,必须单独下载
# 这个时候还在cactus-bin-v2.9.9这个文件夹下
cd bin
# 要给可执行的权限
for i in wigToBigWig faToTwoBit bedToBigBed bigBedToBed axtChain pslPosTarget bedSort hgGcPercent mafToBigMaf hgLoadMafSummary hgLoadChain; do
wget -q http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/${i};
chmod +x ${i};
done
总体参考:
https://github.com/ComparativeGenomicsToolkit/cactus/blob/v2.9.9/BIN-INSTALL.md
如果下载的是源代码或者在docker里面使用可以参考这个:
https://github.com/ComparativeGenomicsToolkit/cactus/blob/v2.9.9/README.md
检测使用:
source venv-cactus-v2.9.9/bin/activate
# 看看能不能找到
which cactus
# 看看版本编号
cactus --version
03 使用方法
1. 输入准备:
1.1 基因组文件:每个样本的FASTA文件(如sample1.fa, sample2.fa)。
1.2 进化树文件(tree.txt):即使同物种也需简单定义分支关系(例:(sample1:1,sample2:1);)。如果是不同种,进化树文件需要反映真实分支关系(例:((human:0.1,mouse:0.1):0.05,zebrafish:0.15);)
2. 运行命令:
cactus jobstore seqFile outputHal
usage: cactus [-h] [--config PATH] [--logCritical] [--logError] [--logWarning] [--logDebug] [--logInfo] [--logOff]
[--logLevel {Critical,Error,Warning,Debug,Info,critical,error,warning,debug,info,CRITICAL,ERROR,WARNING,DEBUG,INFO}] [--logFile LOGFILE][--rotatingLogging]
[--logColors BOOL][--workDir PATH][--coordinationDir PATH][--noStdOutErr][--stats][--clean {always,onError,never,onSuccess}]
[--cleanWorkDir {always,onError,never,onSuccess}][--clusterStats [OPT_PATH]] [--restart]
[--batchSystem {aws_batch,single_machine,grid_engine,lsf,mesos,slurm,torque,htcondor,kubernetes}][--disableHotDeployment]
[--disableAutoDeployment DISABLEAUTODEPLOYMENT][--maxJobs MAX_JOBS][--maxLocalJobs MAX_LOCAL_JOBS][--manualMemArgs][--runLocalJobsOnWorkers]
[--coalesceStatusCalls][--statePollingWait STATEPOLLINGWAIT][--statePollingTimeout STATE_POLLING_TIMEOUT][--batchLogsDir BATCH_LOGS_DIR]
[--awsBatchRegion AWS_BATCH_REGION][--awsBatchQueue AWS_BATCH_QUEUE][--awsBatchJobRoleArn AWS_BATCH_JOB_ROLE_ARN][--scale SCALE]
[--dont_allocate_mem | --allocate_mem][--symlinkImports BOOL][--moveOutputs BOOL][--caching BOOL][--provisioner {aws,gce,None}][--nodeTypes NODETYPES]
[--maxNodes INT[,INT...]] [--minNodes INT[,INT...]] [--targetTime INT][--betaInertia FLOAT][--scaleInterval INT][--preemptibleCompensation FLOAT]
[--nodeStorage INT][--nodeStorageOverrides NODETYPE:NODESTORAGE[,NODETYPE:NODESTORAGE...]] [--metrics BOOL][--assumeZeroOverhead BOOL]
[--maxServiceJobs INT][--maxPreemptibleServiceJobs INT][--deadlockWait INT][--deadlockCheckInterval INT][--defaultMemory DEFAULTMEMORY]
[--defaultCores FLOAT][--defaultDisk INT][--defaultAccelerators ACCELERATOR[,ACCELERATOR...]] [--defaultPreemptible [BOOL]] [--maxCores INT]
[--maxMemory INT][--maxDisk INT][--retryCount INT][--enableUnlimitedPreemptibleRetries BOOL][--doubleMem BOOL][--maxJobDuration INT]
[--rescueJobsFrequency INT][--jobStoreTimeout FLOAT][--maxLogFileSize MAXLOGFILESIZE][--writeLogs [OPT_PATH]] [--writeLogsGzip [OPT_PATH]]
[--writeLogsFromAllJobs BOOL][--writeMessages PATH][--realTimeLogging REALTIMELOGGING][--disableChaining BOOL]
[--disableJobStoreChecksumVerification BOOL][--sseKey PATH][--setEnv NAME=VALUE or NAME][--servicePollingInterval FLOAT][--forceDockerAppliance BOOL]
[--statusWait INT][--disableProgress][--debugWorker][--disableWorkerOutputCapture][--badWorker FLOAT][--badWorkerFailInterval FLOAT]
[--configFile CONFIGFILE][--root ROOT][--latest][--containerImage CONTAINERIMAGE][--binariesMode {docker,local,singularity}][--gpu [GPU]]
[--lastzCores LASTZCORES][--lastzMemory LASTZMEMORY][--consCores CONSCORES][--consMemory CONSMEMORY][--intermediateResultsUrl INTERMEDIATERESULTSURL]
[--skipPreprocessor][--maxOutgroups MAXOUTGROUPS][--chromInfo CHROMINFO]
jobStore seqFile outputHal
其中seqFile是配置文件,里面包含了物种的进化树文件(第一行)、fa序列文件名称以及所在位置。outputHal是输出的HAL文件名称。当分析不同物种时,需要用--root 指定一下根。
04 输出文件及处理
1. 输出文件:outputHal
输出的文件是Hal格式,无法直接进行查看,只能使用halStats做统计,但是cactus提供了很多命令可以进行格式转化。
#统计命令:
halStats outputHal > stats.txt
2. 后续处理
2.1 转换成maf文件:
hal2maf output.hal output.maf
# 推荐参数: -refGenome xxx --chunkSize 500000 --dupeMode single --noAncestors
# 提取时一定要有参考基因组。 --dupeMode single只提取单拷贝; --noAncestors 不包含构建好的祖先序列 chunkSize随便选,大一点结果好,只是大一点提取应该会慢。
# 但是hal2maf很慢,只能处理小文件,所以推荐cactus-hal2maf
2.2 转化成vg文件
hal2vg output.hal --refGenomes XX --noAncestors > output.vg
2.3 转成GBZ
vg autoindex --workflow giraffe -v output.hal -p pangenome -t 32
2.4 生成GFA
vg convert output.hal -g > pangenome.gfa
2.5 生成ODGI
vg convert output.hal -o > pangenome.og
odgi sort -i pangenome.og -o pangenome.sorted.og -P -t 32
参考:
https://www.nature.com/articles/s41586-020-2871-y
https://github.com/ComparativeGenomicsToolkit/cactus/blob/master/doc/progressive.md
- 发表于 2025-09-01 10:03
- 阅读 ( 3084 )
- 分类:基因组学
