LeafCutter可变剪切注释
目前主流的探究转录组数据的可变剪切的算法是基于estimate isoform ratios 或者 exon inclusion levels ,但是可变剪切本跟正常转录本重合的比例很大,存在技术误差也,且他依赖于基因现有的注释信息,既不准确,也不完全,而leafcutter软件可以弥补这些缺陷
01
—
数据准备
首先需要做一个标准的leafcutter分析,得到每个样本的内含子表达量信息
perind_numers.counts.gz这个文件
MPC14L sample1 sample2 sample3 sample4 ...
Chr04:71053:71791:clu_1_NA 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 1 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0
Chr04:71059:71791:clu_1_NA 27 8 24 28 20 48 50 36 16 45 20 16 15 14 20 56 19 44 34 23 44 44 29 30 63 27 35 52 9 20 40 33 17 33 70 35 53 31 53 33 37 41 38 33 51 2
Chr04:71946:72077:clu_2_NA 22 9 17 24 24 26 32 24 18 15 28 23 31 2 48 48 8 31 25 12 66 39 20 24 48 18 46 23 17 24 33 21 31 16 37 37 34 13 43 24 37 31 17 21 40 19
Chr04:71949:72077:clu_2_NA 3 0 3 1 2 0 2 0 0 0 0 0 0 3 0 0 0 0 0 0 0 1 2 1 0 0 0 1 0 3 0 0 1 0 3 4 1 0 0 0 0 0 0 1 1 0 0 0 1 0 1 0 0 0 0 0 0 0 0 2 1 1 0 0 0 0 0
Chr04:72205:72863:clu_3_NA 0 1 0 0 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
里面每一行都是一个内含子,每一列都是一个样本,写明了它们的表达值,这些数值就可以用来做可变剪切分析
随后提供一个样本分组情况,和gtf文件
进行差异分析使用这个脚本leafcutter_ds.R
随后就能得到最关键的文件leafcutter_ds_cluster_significance.txt
格式如下
status loglr df p cluster p.adjust
Success 2.64193838435494 2 0.0712230779654073 chrChr01:clu_19321_NA 0.214269156212565
Success 2.12197496142517 3 0.236298335739971 chrChr01:clu_19322_NA 0.452086256828289
Success 2.92283956715858 2 0.0537807562049738 chrChr01:clu_19323_NA 0.177868987773832
Success 0.682929338673375 1 0.242524696579446 chrChr01:clu_19324_NA 0.458887273884253
<=1 sample with coverage>min_coverage NA NA NA chrChr01:clu_19325_NA NA
Success 12.9546387630635 2 2.36522178344153e-06 chrChr01:clu_19326_NA 3.99620864465738e-05
Success 0.168755118676472 1 0.561269897737211 chrChr01:clu_19327_NA 0.741275092150014
其实就是每一个内含子聚类的差异及显著性,有些没有success的就是覆盖度不够的,没有后续分析的意义了
02
—
绘图
还有一些需要准备的,除了之前生成的文件以外还需要一个样本分组和
一个外显子信息(这个需要从gtf里获取)
这里提供一个小脚本
awk -F'\t' '$3=="exon"{split($9,a,"gene_id \"|\""); print $1"\t"$4"\t"$5"\t"$7"\t"a[2]}' all.final.transcript.gtf | sed 's/";.*//' > exons_simple.txtecho -e "chr\tstart\tend\tstrand\tgene_name" | cat - exons_simple.txt > exons_final.txt
得到的外显子文件格式如下
chr start end strand gene_nameChr01 10414 12178 - Pca01G000010.1Chr01 12277 13890 - Pca01G000010.1Chr01 14058 15364 - Pca01G000010.1Chr01 19142 19460 - Pca01G000020.1Chr01 19540 19596 - Pca01G000020.1Chr01 20145 20210 - Pca01G000020.1Chr01 20287 20376 - Pca01G000020.1Chr01 20495 20575 - Pca01G000020.1Chr01 21408 21512 - Pca01G000020.1Chr01 21603 21738 - Pca01G000020.1Chr01 21875 22697 - Pca01G000020.1Chr01 22823 23314 - Pca01G000020.1
随后绘图
ds_plots.R -e gencode19_exons.txt.gz perind_numers.counts.gz group_info.txt leafcutter_ds_cluster_significance.txt -f 0.05
算的非常快,最后给你一组图片,
ds_plots.pdf
可以直接查看,里面有很多基因的剪切情况,非常直观这个软件默认出30个剪切图片,可以通过-m调整
我这里切一张长图
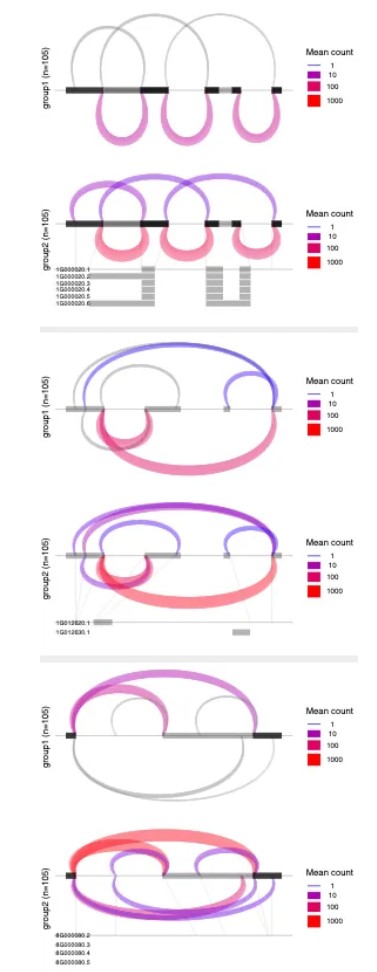
大部分样本虽然剪切形式没有变化,但是不同的转录本表达量差异很大
少部分出现了可变剪切形式,比如最后这个
参考:
https://leafcutter.shinyapps.io/leafviz/
http://www.bio-info-trainee.com/2949.html
http://www.bio-info-trainee.com/5776.html
- 发表于 2026-01-05 17:27
- 阅读 ( 654 )
- 分类:GWAS
