选择清除分析之XP-EHH
在群体遗传学中,选择清除、连锁不平衡和单倍型是三个紧密关联的核心概念,它们共同帮助我们理解自然选择如何在基因组上留下印记。今天先来说说选择清除分析。
一、 核心概念解析
选择清除:是指由于强烈的正选择,使得一个有益突变及其连锁的基因组区域在群体中的遗传多样性显著降低甚至完全消失的现象。就像扫帚扫过一样,清除了该区域的变异。
连锁不平衡:是指不同基因组位点上的等位基因并非独立遗传,而是倾向于共同遗传的现象。当一个有益突变受到正选择时,它不仅自身频率会升高,其周围紧密连锁的基因组区域也会被“搭便车”而一起被固定下来,导致这些区域内的位点间出现高度的连锁不平衡。
单倍型:是指位于一条染色体上的一组紧密连锁、共同遗传的等位基因的组合。它可以被看作是一段DNA序列的“指纹”或特定组合。
这三者之间的关系可以串联成一个完整的叙事,来描述一次选择性事件的发生及其后果:自然选择驱动 -> 选择清除产生 -> 连锁不平衡升高 -> 特定单倍型频率扩张。
二、选择清除分析
选择清除分析的核心原理是:在受选择的基因组区域,会出现有利等位基因频率快速上升、核苷酸多态性降低、连锁不平衡增加等特征。通过检测这些特征,可以识别出可能受到自然选择或人工选择的基因区域。
选择清除分析的四个主要方面:
1. 基于核苷酸多态性降低的方法
这类方法通过检测基因组区域的核苷酸多样性(π值)降低来识别选择信号。主要包括:
π值(核苷酸多样性):衡量群体内遗传多态性,受选择区域π值显著低于基因组背景值
π ratio:比较两个群体的π值比值,用于识别相对受选择区域
ROD(Reduction of Diversity):基于野生群体和栽培群体的核苷酸多样性差异
2. 基于等位基因频率改变的方法
这类方法通过检测等位基因频率的异常变化来识别选择信号:
Tajima's D:中性检验统计量,负值表明可能存在正选择
CLR(复合似然比检验):检测单个群体内的选择信号
XP-CLR(跨群体复合似然比检验):比较两个群体间的等位基因频率差异
3. 基于连锁不平衡增加的方法
这类方法通过检测单倍型纯合度的异常增加来识别选择信号:
EHH(扩展单倍型纯合度):检测单个群体内单倍型纯合度的延伸长度
iHS(整合单倍型得分):标准化后的EHH统计量
XP-EHH(跨群体扩展单倍型纯合度):比较两个群体间的单倍型纯合度差异
4. 基于群体分化的方法
这类方法通过比较不同群体间的遗传分化来识别选择信号:
Fst:群体间固定指数,值越大表示分化程度越高
dXY:群体间核苷酸差异,用于识别高分化区域
三、XP-EHH
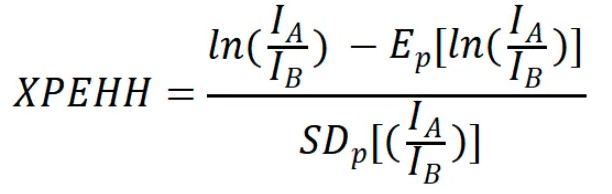
四、Hapbin
1. 软件下载安装:
git clone https://github.com/evotools/hapbin.git
cd hapbin/build/
cmake ../src/
make
hapbin/build/ehhbin -h
Usage: ehhbin --map input.map --hap input.hap --locus id
-h,--helpShow this help
-v,--versionVersion information
-d,--hapHap file
-m,--mapMap file
-l,--locusLocus
-c,--cutoffEHH cutoff value (default: 0.05)
-b,--minmafMinimum allele frequency (default: 0.05)
-s,--scaleGap scale parameter in bp, used to scale gaps > scale parameter as in Voight, et al.
-e,--max-extendMaximum distance in bp to traverse when calculating EHH (default: 0 (disabled))
-a,--binomUse binomial coefficients rather than frequency squared for EHH
2. 数据准备:
cat cultivated_popid.txt wild_popid.txt > popid.txt
for i in Chr1 Chr2 ;do
vcftools --vcf clean.vcf --recode --recode-INFO-all --stdout --keep popid.txt --chr ${i} > ${i}.vcf
done
for i in cultivated wild ;do
vcftools --vcf Chr1.vcf --recode --keep ${i}_popid.txt --out ${i}_Chr1;
vcftools --vcf Chr2.vcf --recode --keep ${i}_popid.txt --out ${i}_Chr2;
vcftools --vcf ${i}_Chr1.recode.vcf --IMPUTE --out ${i}_Chr1;
vcftools --vcf ${i}_Chr2.recode.vcf --IMPUTE --out ${i}_Chr2;
done
# 从VCF生成map文件
for i in Chr1 Chr2 ;do
vcftools --vcf ${i}.vcf --plink --out ${i} ;
done
# 转换物理位置为遗传距离
for i in Chr1 Chr2 ;do
awk -v a=${i} '{ print a,$2,$4/1000000,$4}' ${i}.map > ${i}.genetic.map ;
done
# 使用:
xpehhbin --hapA [Population A .hap file]] --hapB [Population B .hap file]] --map [.map file] --out [output prefix]
# 实际运行:
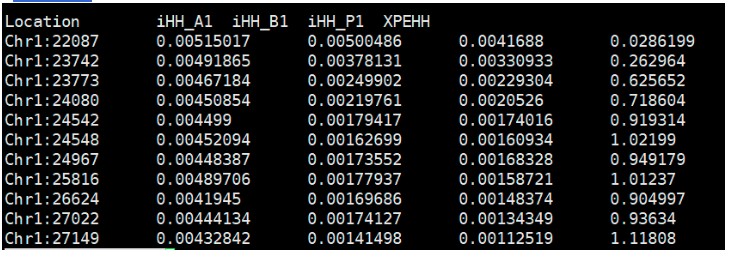
xpehhbin --hapA wild_Chr1.impute.hap --hapB cultivated_Chr1.impute.hap --map Chr1.genetic.map --out result_Chr1
# 也可以写循环分染色体运行
- 发表于 2026-01-04 09:54
- 阅读 ( 1602 )
- 分类:遗传进化
