NCBI下载的10X 空间转录组数据如何读入cellranger or spaceranger
spaceranger
10x的数据可以使用 cellranger mkfastq 产生,下面的例子是两个样本同一个lane测序拆分后分析;第二个例子是同一个样本两个lane测序,需要合并一起分析;
大家注意文件命名方式,蓝色和红色部分;
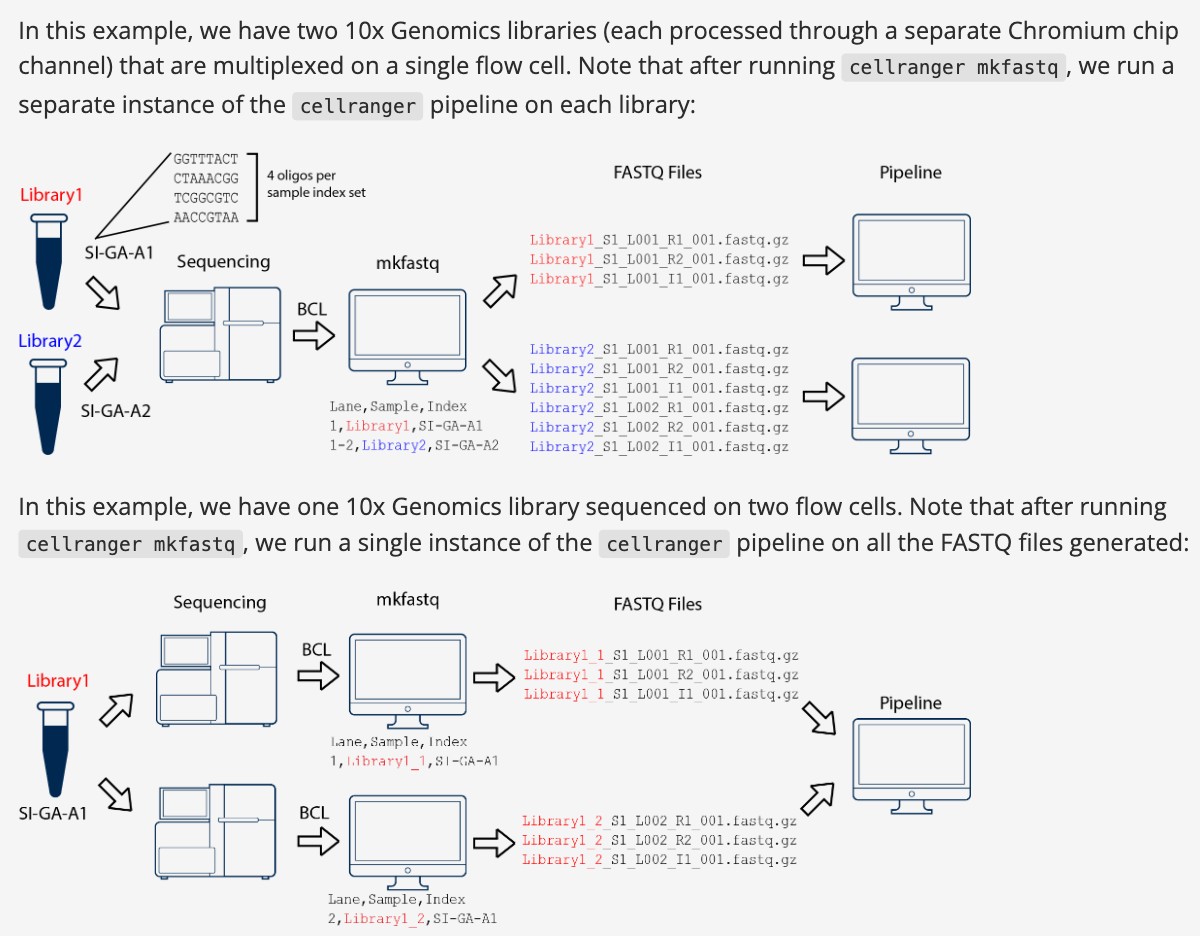
spaceranger命名规则
[Sample Name]_S1_L00[Lane Number]_[Read Type]_001.fastq.gz
# 其中Read Type
# I1: Sample index read (optional)
# R1: Read 1
# R2: Read 2
NCBI下载的数据,为双端reads,不符合spaceranger mkfastq出来的命名规则,后续spaceranger count 无法读入,因此我们要按上述命名规则重新命名文件:fastq文件夹
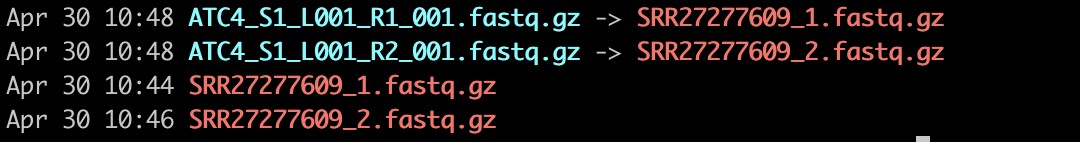
空间图像信息,放在spatial文件夹:
spaceranger读人数据分析;
spaceranger count --id=ATC4 --transcriptome $refdir \ --fastqs=./GSE250521_Thyroid/fastq/ --sample=ATC4 \ --unknown-slide=visium-1 --localcores=20 --create-bam=true \ --image=./GSE250521_Thyroid/ATC4/spatial/tissue_hires_image.png \ --output-dir ATC4
参数说明:
# --id: 输出文件夹名
# --transcriptome:参考基因组的路径
# --probe-set: 探针文件的路径
# --fastqs:fastqs文件的路径
# --sample: 样本的名称,与fastq文件一致
# --cytamage: CytAssist 的图片的路径
# --image:显微镜图片的路径,TIFF或者JPEG格式
# --slide: 玻片的ID , 不知道 使用 --unknown-slide instead: visium-1|visium-2|visium-2-large|visium-hd> :https://www.10xgenomics.com/cn/support/software/space-ranger/latest/getting-started/space-ranger-glossary
# --area: 样本所在玻片上的位置编号
# --loupe-alignment:图片校准的json文件的的路径
# --localcores:CPU核数
# --localmem:最大内存
- 发表于 2025-05-07 13:28
- 阅读 ( 3127 )
- 分类:转录组
