Minimap2安装与使用
minimap2是生信大牛Heng Li在2018年发表的三代序列比对工具。与传统的bwa相比,Minimap2可以用于三代测序, 也支持 splicing awared 比对;与一些传统三代比对工具相比,Minimap2 速度非常快,...
minimap2是生信大牛Heng Li在2018年发表的三代序列比对工具。与传统的bwa相比,Minimap2可以用于三代测序, 也支持 splicing awared 比对;与一些传统三代比对工具相比,Minimap2 速度非常快,而且用法简单,比对结果比较不错,可谓是三代序列处理一大利器。主要的缺点就是耗内存,是牺牲内存换速度,二者不可兼得。
官方说明:
https://github.com/lh3/minimap2#general
https://lh3.github.io/minimap2/minimap2.html#10
一、Minimap2的安装
#conda安装
conda create -n Minimap2
source activate Minimap2
conda install -c bioconda minimap2
#编译安装
wget https://github.com/lh3/minimap2/archive/refs/tags/v2.20.tar.gz
tar -zxvf minimap2-2.24.tar.gz
cd /minimap2-2.24
make
二、Minimap2的使用
- Minimap2 是一个通用的序列比对程序,同时适用于二代和三代数据,可将 DNA 或 mRNA 序列与大型参考数据库进行比对。
- 典型用例包括:
- 1、将 PacBio 或 Oxford Nanopore 基因组读数映射到基因组
- 2、发现长读(long reads)之间的重叠,错误率高达~15%
- 3、PacBio Iso-Seq / Nanopore cDNA / Direct RNA reads与参考基因组的剪接感知比对(long reads)
- 4、Illumina的单端或双端reads对齐
- 5、组装水平到组装水平的对齐
- 6、两个密切相关的物种之间的全基因组比对,差异低于~15%
(一)用法概括
# 有对照参考基因组的长序列
minimap2 -a test/MT-human.fa test/MT-orang.fa > test.sam
# 首先创建一个索引,然后比对
minimap2 -x map-ont -d MT-human-ont.mmi test/MT-human.fa
minimap2 -a MT-human-ont.mmi test/MT-orang.fa > test.sam
# 不同类型数据的使用参数
minimap2 -ax map-pb ref.fa pacbio.fq.gz > aln.sam # PacBio CLR genomic reads
minimap2 -ax map-ont ref.fa ont.fq.gz > aln.sam # Oxford Nanopore genomic reads (ONT)
minimap2 -ax map-hifi ref.fa pacbio-ccs.fq.gz > aln.sam # PacBio HiFi/CCS genomic reads (v2.19 or later)
minimap2 -ax asm20 ref.fa pacbio-ccs.fq.gz > aln.sam # PacBio HiFi/CCS genomic reads (v2.18 or earlier)
minimap2 -ax sr ref.fa read1.fa read2.fa > aln.sam # short genomic paired-end reads
minimap2 -ax splice ref.fa rna-reads.fa > aln.sam # spliced long reads (strand unknown)
minimap2 -ax splice -uf -k14 ref.fa reads.fa > aln.sam # noisy Nanopore Direct RNA-seq
minimap2 -ax splice:hq -uf ref.fa query.fa > aln.sam # Final PacBio Iso-seq or traditional cDNA
minimap2 -ax splice --junc-bed anno.bed12 ref.fa query.fa > aln.sam # prioritize on annotated junctions
minimap2 -cx asm5 asm1.fa asm2.fa > aln.paf # intra-species asm-to-asm alignment
minimap2 -x ava-pb reads.fa reads.fa > overlaps.paf # PacBio read overlap
minimap2 -x ava-ont reads.fa reads.fa > overlaps.paf # Nanopore read overlap
(二)基础用法
#1、在基础用法中,minimap2 将ref和query作为输入并生成近似映射(mapping),没有基本级别的对齐(base-level alignment),即坐标只是近似的,且输出中没有 CIGAR
minimap2 ref.fa query.fq > approx-mapping.paf
#2、在PAF文件中制造CIGAR的cg标签
minimap2 -c ref.fa query.fq > alignment.paf
#3、以SAM 格式输出对齐
minimap2 -a ref.fa query.fq > alignment.sam
#4、建立索引来进行比对
minimap2 -d ref.mmi ref.fa # indexing
minimap2 -a ref.mmi reads.fq > alignment.sam # alignment
(三)三代长reads的基因组比对
#Map long noisy genomic reads(长噪声读取)
minimap2 -ax map-pb ref.fa pacbio-reads.fq > aln.sam # for PacBio CLR reads
minimap2 -ax map-ont ref.fa ont-reads.fq > aln.sam # for Oxford Nanopore reads
# -a:设置输出为sam格式
#-x:对不同类型数据,设置不同参数
(四)查找长reads之间的重叠
minimap2 -x ava-pb reads.fq reads.fq > ovlp.paf # PacBio CLR read overlap
minimap2 -x ava-ont reads.fq reads.fq > ovlp.paf # Oxford Nanopore read overlap
(五)二代短reads的基因组比对
minimap2 -ax sr ref.fa reads-se.fq > aln.sam # single-end alignment
minimap2 -ax sr ref.fa read1.fq read2.fq > aln.sam # paired-end alignment
minimap2 -ax sr ref.fa reads-interleaved.fq > aln.sam # paired-end alignment
#作者在github介绍说Minimap2不适用于短拼接读取(short spliced reads)
(六)全基因组之间或组装水平的比对
minimap2 -ax asm5 ref.fa asm.fa > aln.sam # assembly to assembly/ref alignment
#asm5/asm10/asm20:asm到ref映射,用于约0.1/1/5%的序列差异
#对于跨物种全基因组比对,评分系统需要根据序列分歧进行调整
三、Minimap2的输出
(一).paf结果:包括核心的12列字段以及其他
##核心12列字段##
Col Description
1 Query序列ID
2 Query序列长度
3 Query比对开始位置(*based)
4 Query比对结束位置(*based)
5 char,如果query/target是正链关系,'+'表示;负链关系,'-'表示
6 Target序列ID
7 Target序列长度
8 Target比对开始位置
9 Target结束开始位置
10 比对上的碱基数 (matching bases)
11 对齐区域长度(包括gaps)
12 比对质量(0-255)
##其他补充信息##
tp aln类型:P/primary, S/secondary and I,i/inversion
cm Number of minimizers on the chain
s1 Chaining score
s2 Chaining score of the best secondary chain
NM otal number of mismatches and gaps in the alignment
MD To generate the ref sequence in the alignment(在比对过程中生成参考序列)
AS DP alignment score
SA List of other supplementary alignments(其他补充比对列表)
ms DP score of the max scoring segment in the alignment
nn Number of ambiguous bases in the alignment(对齐中不明确的碱基数)
ts Transcript strand (splice mode only)
cg CIGAR string (only in PAF)
cs Difference string
dv Approximate per-base sequence divergence
de Gap-compressed per-base sequence divergence
rl Length of query regions harboring repetitive seeds
(二). SAM结果:SAM文件由两部分组成,头部区和主体区,都以tab分列
#头部区:以’@'开始,体现了比对的一些总体信息。比如比对的SAM格式版本,比对的参考序列,比对使用的软件等
#主体区:比对结果,每一个比对结果是一行,有11个主列和一个可选列
列 关键字 描述
1 QNAME Query序列ID
2 FLAG Bwise FLAG(表示比对类型:paring,strand,mate strand等),如:0,99,256,2048等
3 RENAME Target序列ID
4 POS 比对到参考序列上的位置,从1开始计数;未比对上为0
5 MAPQ 比对的质量分数(越高说明比对到参考序列的上的位置越准确)。如果是255,说明该比对值无效。
6 CIGAR 简要比对信息表达式
7 MRNM 下一片段比对上的参考序列编号
8 MPOS 下一片段比对上的位置,如果不可用,此处为0
9 ISIZE 插入片段长度
10 SEQ 和参考序列在同一个链上比对的序列(若比对结果在负义链上,则序列是其反向重复序列,反向互补序列)
11 QUAL 比对序列的质量(ASCII-33=Phred base quality)reads碱基质量值
12 Optional Fields 可选的列以TAG:TYPE:VALUE的形式提供额外的信息
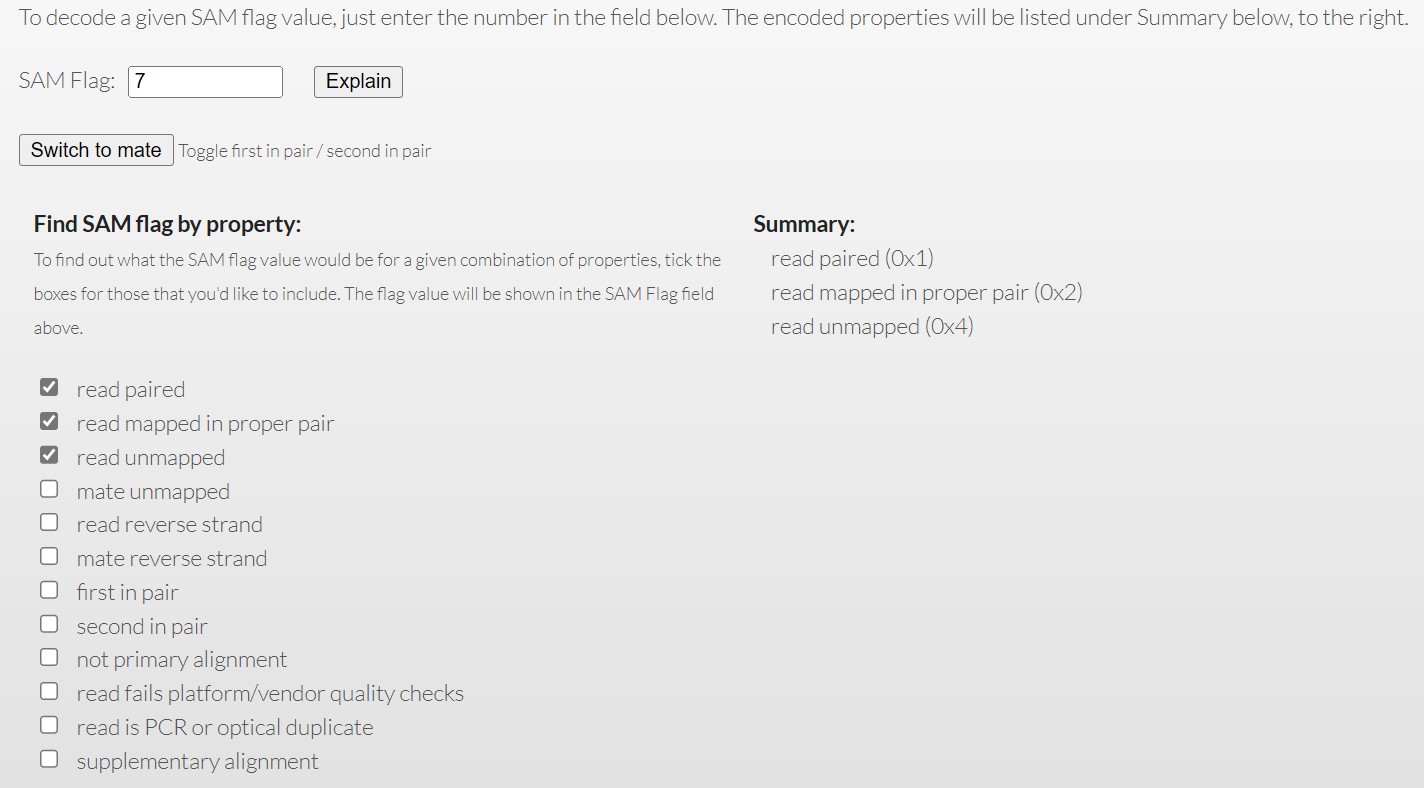
(三)FLAG释义和CIGAR string
1、FLAG释义,常见于bam文件中
1 : 代表这个序列采用的是PE双端测序
2: 代表这个序列和参考序列完全匹配,没有插入缺失
4: 代表这个序列没有mapping到参考序列上
8: 代表这个序列的另一端序列没有比对到参考序列上,比如这条序列是R1,它对应的R2端序列没有比对到参考序列上
16:代表这个序列比对到参考序列的负链上
32 :代表这个序列对应的另一端序列比对到参考序列的负链上
64 : 代表这个序列是R1端序列, read1;
128 : 代表这个序列是R2端序列,read2;
256: 代表这个序列不是主要的比对,一条序列可能比对到参考序列的多个位置,只有一个是首要的比对位置,其他都是次要的
512: 代表这个序列在QC时失败了,被过滤不掉了(# 这个标签不常用)
1024: 代表这个序列是PCR重复序列(#这个标签不常用)
2048: 代表这个序列是补充的比对(#这个标签具体什么意思,没搞清楚,但是不常用)
官方对flag的解释:Explain SAM Flags (broadinstitute.github.io) 可以根据自己的需求检测到对应的参数,后续可用samtools进行筛选。
2、CIGAR string
- CIGAR string ,简要比对信息表达式(Compact Idiosyncratic Gapped AlignmentReport),其以参考序列为基础,使用数字加字母表示比对结果,比如3S6M1P1I4M,前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的,字母的含义如下:

参考:https://www.jianshu.com/p/d1868194b65e
- 发表于 2023-05-12 11:07
- 阅读 ( 16567 )
- 分类:软件工具
