TCGA SNV 体细胞突变下载
TCGA SNV 体细胞突变下载
###############################################################################################
##########加载需要的包 ,包不存在就安装
#############################################################
package_list <- c("TCGAbiolinks","tidyverse","maftools")
for(p in package_list){
if(!suppressWarnings(suppressMessages(require(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))){
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(p)
suppressWarnings(suppressMessages(library(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))
}
}
query <- GDCquery(
project = "TCGA-STAD",
data.category = "Simple Nucleotide Variation",
data.type = "Masked Somatic Mutation",
access = "open"
)
GDCdownload(query )
# 保存整理下载数据结果
maf.data <- GDCprepare(query )
write.table(data.frame(maf.data,check.names = F), file ='maf.tsv', sep="\t",row.names =F, quote = F)
######################################################################
# maftools plot
#######################################################################
selcol=c("Hugo_Symbol", "Chromosome", "Start_Position", "End_Position", "Reference_Allele", "Tumor_Seq_Allele2", "Variant_Classification", "Variant_Type" , "Tumor_Sample_Barcode")
maftools_df=maf.data[,selcol]
write.table(data.frame(maftools_df,check.names = F), file = paste0(opt$outdir,"/",opt$project,'_maftools_df.maf'), sep="\t",row.names =F, quote = F)
maf = read.maf(maf =paste0(opt$outdir,"/",opt$project,'_maftools_df.maf') )
pdf("maf_tmb.pdf",w=8,h=8)
#计算TMD
maf.tmd = tmb(maf = maf,
captureSize = 50,
logScale = TRUE)
maf.tmd<-as.data.frame(maf.tmd)
head(maf.tmd)
dev.off()
a<-t(as.data.frame(strsplit(as.character(maf.tmd$Tumor_Sample_Barcode),"-")))
patientID<-paste0(a[,1],"-",a[,2],"-",a[,3])
write.table(data.frame(maf.tmd,patient=patientID),file="tmb.tsv",sep="\t",quote = F,row.names = F)
pdf("maf_plot.pdf",w=5,h=5)
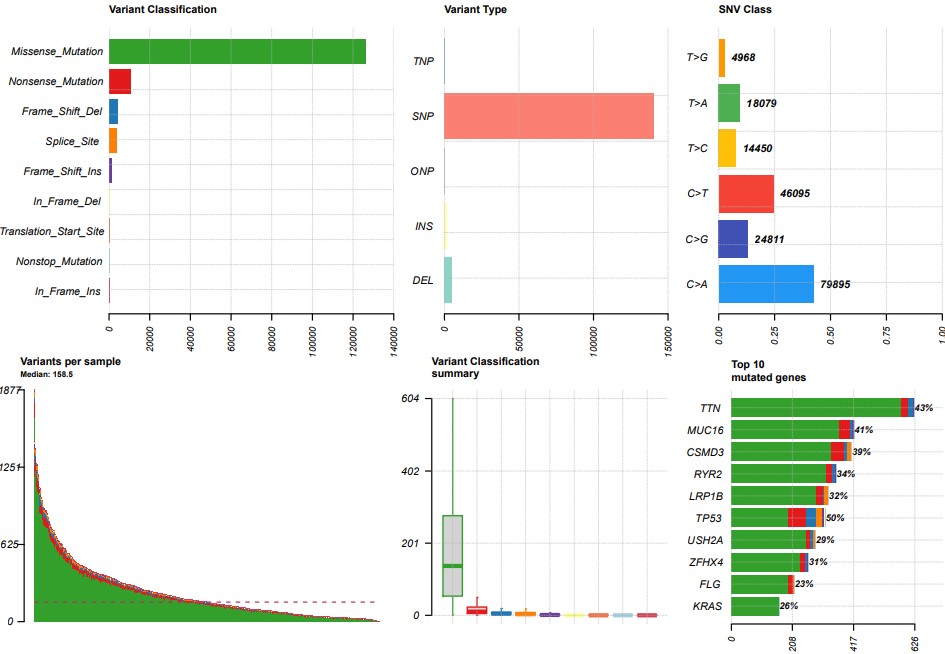
plotmafSummary(maf = maf, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE,titvRaw = FALSE)
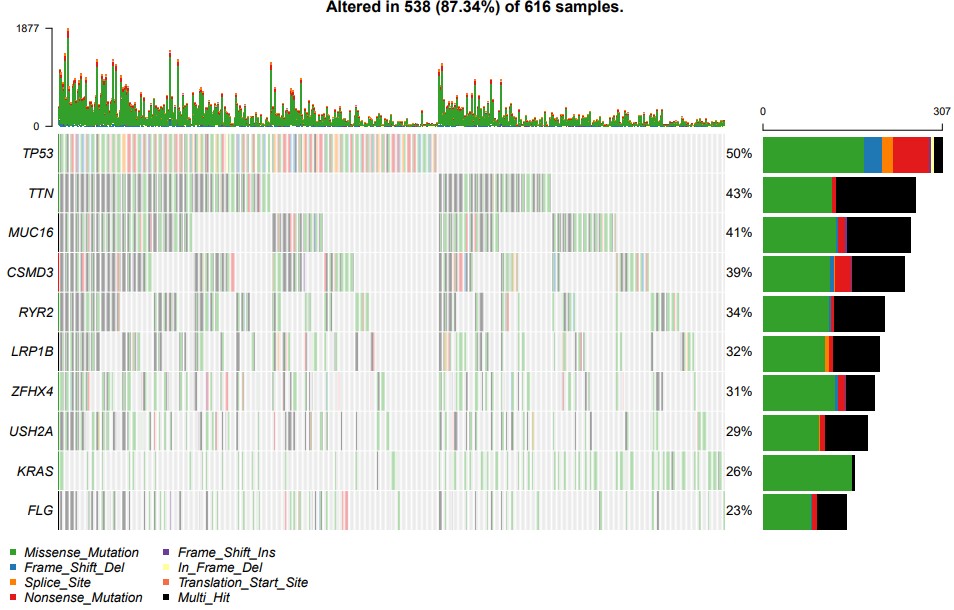
oncoplot(maf = maf, top = 10)
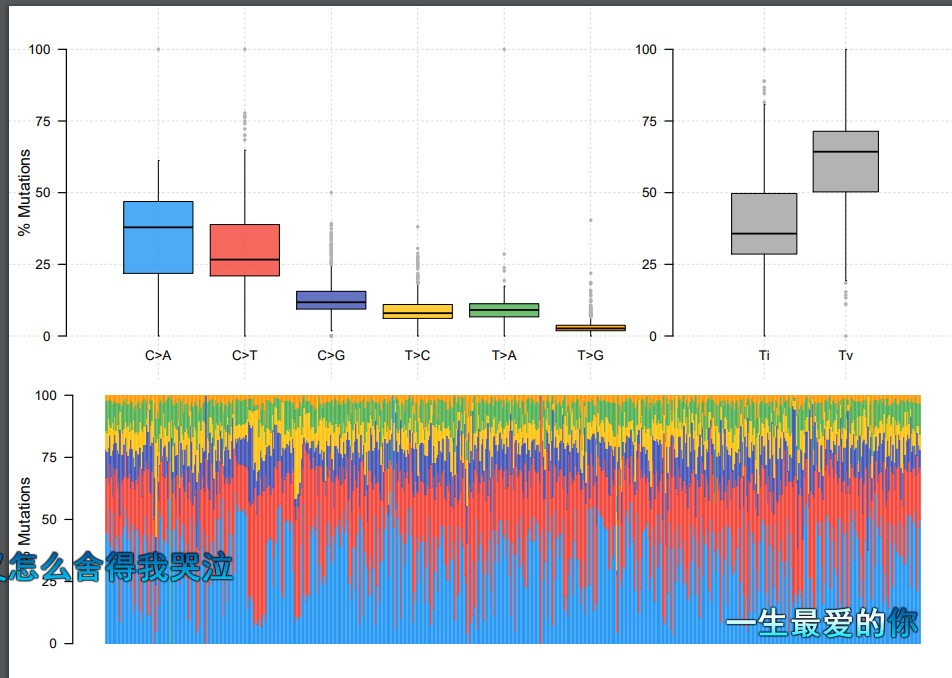
titv = titv(maf = maf, plot = FALSE, useSyn = TRUE)
#plot titv summary
plotTiTv(res = titv)
dev.off()
|sort(harmonized.data.type) |
|:-----------------------------------|
|Aggregated Somatic Mutation |
|Aligned Reads |
|Allele-specific Copy Number Segment |
|Annotated Somatic Mutation |
|Biospecimen Supplement |
|Clinical Supplement |
|Copy Number Segment |
|Differential Gene Expression |
|Gene Expression Quantification |
|Gene Level Copy Number |
|Gene Level Copy Number Scores |
|Isoform Expression Quantification |
|Masked Copy Number Segment |
|Masked Intensities |
|Masked Somatic Mutation |
|Masked Somatic Mutation |
|Methylation Beta Value |
|miRNA Expression Quantification |
|Protein Expression Quantification |
|Protein Expression Quantification |
|Raw CGI Variant |
|Raw Simple Somatic Mutation |
|Single Cell Analysis |
|Slide Image |
|Splice Junction Quantification |
- 发表于 2022-08-22 20:39
- 阅读 ( 3671 )
- 分类:TCGA
