envfit()的线性拟合:非约束排序(PCA、CA、PCoA、NMDS)
非约束排序(PCA、CA、PCoA、NMDS)中被动拟合解释变量在R中的实现方法在群落排序分析中,若我们想在排序图中尽量展示最大的物种矩阵的变差(variation)的同时,又想展示出环境变量(即解释变量);或者当我们在约束排序(如使用RDA、CCA等)中发现环境变量的解释程度偏低,约束排序模型不是很适用时;或者发现同时使用了物种矩阵和环境变量矩阵的约束排序结果较为混乱,各组样方之间不易区分,相较之下只使用物种矩阵的非约束排序的效果却很理想,但是我们还想在其中添加环境变量作为解释变量。这些情形下,不妨考虑试一下在非约束排序模型(PCA、CA、PCoA、NMDS等)中被动拟合环境变量的方式。本篇将使用R包vegan中的一些列函数命令,简介这种方法的执行过程。对于该方法涉及到的基础概念原理,可参考前文。
示例文件、R脚本等,已上传至百度盘,提取码:0v4g。https://pan.baidu.com/s/18Nq6-kJUgJiCr_BV3qAtRQ
示例数据说明
某研究对三种环境中的土壤进行了采样(环境A、B、C,各环境下采集9个土壤样本),结合二代测序观测了土壤细菌群落物种多度,并对多种土壤理化指标进行了测量,获得以下数据。
文件“phylum_table.txt”为通过16S测序所得的细菌类群丰度表格,此处统计至细菌“门水平”(即界门纲目科属种的“门”这一水平)。第一列为各样本的名称,之后每一列为一类门,交叉区域为每类门在各样本中的丰度。
备注:本篇中为了便于操作,直接使用了门水平的丰度表,并未使用OTU水平等更细致层级的丰度表作演示。且本篇仅对方法(R代码)作展示,不探讨数据的合理性或结果生态学意义等。在实际的数据分析中,需注意。
文件“env_table.txt”记录了各样本所在采样环境的多种土壤理化指标信息。第一列为各样本的名称,之后每一列为一种土壤理化因子;交叉区域为各采样样本所处环境中,各土壤理化指标的测量数值。
备注:本文中为了便于操作,所使用的理化因子均为数值型变量,未使用到因子类型的变量(分类变量、定性变量)。各土壤理化因子的描述可见网盘附件“附:env说明.txt”
在下文为了方便描述,将这里的“细菌门”直接称为“物种”(尽管并非“种”分类层级),因此“细菌门水平丰度组成”也直接称为“物种多度”;同时也将“土壤理化”统称为“环境变量”;为了方便与vegan中的名词对应,将“样本”统称为“样方”。
接下来首先对物种多度数据执行非约束排序(PCA、CA、PCoA、NMDS),观察各样方在排序空间中的位置(群落物种多度相似程度),以及各物种(或主要物种)在各样方中的丰度分布规律。然后再通过多元回归的方式将环境变量被动地添加到非约束轴中,观察各样方所处的环境梯度,并使用环境变量对群落组成及物种分布作出解释。
envfit()的线性拟合
首先将物种多度数据以及环境变量数据读到R中,然后加载vegan包。
vegan中,可通过envfit()函数实现在非约束轴中被动拟合解释变量的过程,它是一种线性的方法。由于envfit()只能与vegan包中的排序函数一起配合使用,下文对四种常见的非约束排序(PCA、CA、PCoA、NMDS)都简介一下。
#加载 vegan 包
library(vegan)
#读入物种多度数据
phylum <- read.delim('phylum_table.txt', row.names = 1, sep = '\t', stringsAsFactors = FALSE, check.names = FALSE)
#读取环境数据
env <- read.delim('env_table.txt', row.names = 1, sep = '\t', stringsAsFactors = FALSE, check.names = FALSE)
主成分分析(PCA)中的方法
以此为例详细展示细节。
PCA排序
首先执行PCA,在vegan中,PCA使用rda()执行。由于物种多度数据中通常含有很多0值,推荐在PCA前进行Hellinger转化后使用(此时即为tb-PCA)。详情参考前文PCA。
##tb-PCA
#物种数据 Hellinger 预转化
phylum_hel <- decostand(phylum, method = 'hellinger')
#PCA 排序
tbpca <- rda(phylum_hel, scale = FALSE)
#查看 PCA 结果等,I 型标尺为例
summary(tbpca, scaling = 1)
plot(tbpca, choices = c(1, 2), scaling = 1, display = c('wa'))
拟合环境变量至PCA轴
接下来使用envfit()将环境变量以多元回归的方式拟合至PCA非约束轴中,原理可可参考前文。
#环境变量与非约束轴的多元回归,999 次置换检验,详情 ?envfit()
tbpca_ef <- envfit(tbpca~., data = env, perm = 999, choices = c(1,2), display = 'sites')
tbpca_ef
式中“tbpca~.”意为将环境变量矩阵env中的所有列(各类环境变量)拟合至PCA轴;默认使用choices = c(1,2),拟合至前两轴(本示例不关注第三及以后的PCA轴);display参数,将环境变量与样方得分(sites,即PCA结果中的样本排序坐标)或物种得分(lc,即PCA结果中的物种排序坐标)进行拟合,这里使用display = 'sites'(对于回归本身而言,拟合非约束轴中的样方得分或物种得分均可以,实际的数据分析中视情况考虑使用哪种);perm = 999,随机置换999次以检验各环境变量的显著性。
“tbpca_ef”的结果如下所示。与RDA相似,环境变量投影到排序图中以向量的形式作为展示,下图中PC1和PC2即分别为各环境变量向量的顶点在PCA第一轴和第二轴中的坐标;r2为多元回归的回归决定系数R2;Pr(>r)即显著性p值。
示例中共给定了9个环境变量,将它们与PCA前两轴中的样方得分进行多元回归,并通过999次置换检验后,得到其中的6个环境变量与样方得分是具有显著的线性关系的。
置换检验的p值校正
为了减少I类错误,必要时可考虑对置换检验的p值作校正,原理可参考前文。
如下示例使用了Bonferroni校正方法。
#p 值校正,bonferroni 方法
tbpca_ef_adj <- tbpca_ef
tbpca_ef_adj$vectors$pvals <- p.adjust(tbpca_ef_adj$vectors$pvals, method = 'bonferroni')
tbpca_ef_adj
p值校正后,对于上述示例,最终仅保留了4个显著的环境变量。而且我们发现,这4个环境变量与PCA非约束轴样方得分之间存在较为可观的线性关系(根据R2判断)。
主要结果提取
如有需要,将PCA排序的样方得分、物种得分、投影的环境变量得分等提取出。
#提取样方得分(坐标),I 型标尺为例,前两轴
tbpca_site.scaling1 <- scores(tbpca, choices = 1:2, scaling = 1, display = 'sites')
#或者
tbpca_site.scaling1 <- summary(tbpca, scaling = 1)$sites[ ,1:2]
#若输出在本地,以 csv 格式为例
write.csv(data.frame(tbpca_site.scaling1), 'pca_site.scaling1.csv', quote = FALSE)
#提取物种得分(坐标),I 型标尺为例,前两轴
tbpca_species.scaling1 <- summary(tbpca, scaling = 1)$species[ ,1:2]
#若输出在本地,以 csv 格式为例
write.csv(data.frame(tbpca_species.scaling1), 'pca_species.scaling1.csv', quote = FALSE)
#提取特征根、各轴解释量(各轴特征根占总特征根的比值)
eig <- tbpca$CA$eig
eig_prop <- tbpca$CA$eig / sum(tbpca$CA$eig)
#将环境变量的坐标、多元回归 r2、回归显著性(校正后的 p 值)输出在本地,以 csv 格式为例
tbpca_env <- data.frame(cbind(tbpca_ef_adj$vectors$arrows, tbpca_ef_adj$vectors$r, tbpca_ef_adj$vectors$pvals))
names(tbpca_env) <- c('PC1', 'PC2', 'r2', 'p.adj')
write.csv(tbpca_env, 'pca_env.csv', quote = FALSE)
环境变量与样方得分的相关性
环境变量的排序坐标(即排序图中所示的环境向量顶点坐标),由这些环境变量与非约束轴相关性依据R2的平方根缩放后所得,使得拟合R2“较强”的环境变量比“较弱”的环境变量具有更长的向量(箭头)长度。
在本示例中,由于使用的PCA轴的样方得分与环境变量进行的拟合,因此若我们将环境变量排序坐标提取出,并乘以R2的平方根后,即可得到环境变量与样方得分的相关性。
#计算环境变量与样方得分的相关性(只计算显著的 4 个环境变量)
tbpca_env_cor <- cor(env[ ,c('DOC', 'AP', 'AK', 'NH4')], tbpca_site.scaling1, method = 'pearson')
#或者由排序坐标和 r2 反向推算出环境变量与样方得分的相关性
arrow_heads <- tbpca_ef_adj$vectors$arrows #提取 env 向量(坐标)矩阵
r2 <- tbpca_ef_adj$vectors$r #提取 env r2
arrow_heads * sqrt(r2) #即可得相关性,结果和上述“tbpca_env”是一致的
排序图绘制
排序结果最终以排序图的形式呈现出。若我们发现环境变量与非约束轴之间存在较为可观的线性关系,且排序结果也具有良好的生态学意义时,就可以将它们的关系绘制出来整理结果了。
使用vegan打包好的plot()绘制方式展示,以上述tb-PCA示例为例。
#可使用 ?plot.cca 了解作图命令的更多参数信息,注意不要使用 ?plot
#简要作图展示 PCA 结果(仅展示样方排序,不展示物种排序)
plot(tbpca, choices = c(1, 2), scaling = 1, type = 'n')
points(tbpca, choices = c(1, 2), scaling = 1, display = 'sites', pch = 20, col = c(rep('red', 9), rep('orange', 9), rep('green3', 9)))
#作图展示被动拟合环境变量后的 PCA 结果,只保留 p.adj < 0.05 的环境变量
plot(tbpca_ef_adj, choices = c(1, 2), p.max = 0.05)
结果如下所示。若觉得箭头过长,可在作图时使用arrow.mul参数调节(在生态学数据的排序分析中,代表“解释变量”的向量箭头长度通常是允许同比例放大或缩小的,可参见文末“常见问题与说明”;在前述PCA中,也同样对代表“响应变量”的向量箭头作过类似的描述)。
或者将排序坐标提取出后,使用ggplot2等作图,如下示例。
##ggplot2 作图示例
library(ggplot2)
#提取样方和环境变量排序坐标,前两轴,I 型标尺
tbpca_site.scaling1 <- data.frame(summary(tbpca, scaling = 1)$sites[ ,1:2])
tbpca_env_sign <- tbpca_env[which(tbpca_env$p.adj < 0.05),1:2]
#PCA 前两轴贡献度(%)
tbpca_eig <- round(100 * tbpca$CA$eig[1:2] / sum(tbpca$CA$eig), 2)
#读取样本分组数据(附件“group.txt”,第一列为各样本名称,第二列为各样本所对应的分组)
group <- read.delim('group.txt', sep = '\t', stringsAsFactors = FALSE, check.names = FALSE)
#合并样本分组信息,构建 ggplot2 作图数据集
tbpca_site.scaling1$sample <- rownames(tbpca_site.scaling1)
tbpca_site.scaling1 <- merge(tbpca_site.scaling1, group, by = 'sample')
tbpca_env_sign$sample <- NA
tbpca_env_sign$group <- rownames(tbpca_env_sign)
#ggplot2 作图
library(ggplot2)
ggplot(tbpca_site.scaling1, aes(PC1, PC2)) +
geom_point(aes(color = group)) +
scale_color_manual(values = c('red', 'orange', 'green3')) +
theme(panel.grid = element_blank(), panel.background = element_rect(color = 'black', fill = 'transparent'), legend.title = element_blank(), legend.key = element_rect(fill = 'transparent')) +
labs(x = paste('PCA1: ', tbpca_eig[1], '%'), y = paste('PCA2: ', tbpca_eig[2], '%')) +
geom_vline(xintercept = 0, color = 'gray', size = 0.5) +
geom_hline(yintercept = 0, color = 'gray', size = 0.5) +
geom_segment(data = tbpca_env_sign, aes(x = 0,y = 0, xend = PC1/5, yend = PC2/5), arrow = arrow(length = unit(0.2, 'cm')), size = 0.5, color = 'blue') +
geom_text(data = tbpca_env_sign, aes(PC1/5 * 1.1, PC2/5 * 1.1, label = group), color = 'blue', size = 3)
结果如下所示。与上述一致,若觉得箭头长度与整张图片大小比例不合适,可同比例放大或缩小所有箭头的长度后再展示。
CA、PCoA、NMDS等中的方法
和上述 PCA一致,我们也可以使用envfit()将环境变量投影至CA、PCoA、NMDS等非约束排序模型中,以期通过这些环境变量用来对非约束轴作出解释。同样地,envfit()只能与vegan中的CA、PCoA、NMDS函数配合使用。
对应分析(CA)中的方法
在vegan中,CA使用cca()执行,详情参考前文CA。
ca <- cca(phylum)
投影环境变量的过程和上述 tb-PCA 相似,演示过程略(envfit()操作方法一致,注意统计细节上有所差别)。
主坐标分析(PCoA)中的方法
在vegan中,PCoA使用cmdscale()执行,详情参考前文PCoA。
##PCoA 排序
#计算相异矩阵,以 Bray-curtis 距离为例
bray <- vegdist(phylum, method = 'bray')
#PCoA 排序
pcoa <- cmdscale(bray, k = (nrow(phylum) - 1), eig = TRUE)
#被动添加环境变量的过程和上述 tb-PCA 类似,简要演示
#环境变量与非约束轴的多元回归,999 次置换检验
pcoa_ef <- envfit(pcoa~., data = env, perm = 999, choices = c(1,2), display = 'sites')
#p 值校正
pcoa_ef$vectors$pvals <- p.adjust(pcoa_ef$vectors$pvals, method = 'bonferroni')
#PCoA 作图
plot(scores(pcoa)[ ,1], scores(pcoa)[ ,2], pch = 20, col = c(rep('red', 9), rep('orange', 9), rep('green3', 9)))
#将 p.adj < 0.05 的结果投影至 PCoA 排序图
plot(pcoa_ef, choices = c(1, 2), p.max = 0.05)
在本示例中,通过置换检验以及p值校正后并未发现存在任何环境变量与PCoA轴存在显著的线性关系。当然并不一定说数据本身不好,也有可能是选择的距离测度不合适。在实际的数据分析中,碰到这种情况时需要仔细思考,例如更换距离矩阵再行尝试。
或者,不再基于样方得分,而是尝试使用物种得分与环境变量进行多元回归拟合(参数display = 'lc')。尽管物种信息在相异矩阵的计算过程中丢失,但是物种得分仍然可以通过物种变量与它们所在样方得分的多度加权平均与PCoA轴建立关联而得到并投影到最终的排序图中。物种变量的投影原理和这里环境变量的投影原理有几分相像。vegan中,在PCoA中投影物种得分的功能可以通过wascores()实现,详情参考前文PCoA。
非度量多维尺度分析(NMDS)中的方法
在vegan中,NMDS使用metaMDS()执行,详情参考前文NMDS。
尽管与PCA、CA、PCoA等基于基于特征向量的排序方法相比,NMDS是一种非参数的方法,但也仍然可以通过envfit()拟合环境变量(可参考文末“常见问题与说明”)。
##NMDS 排序,以 Bray-curtis 距离为例(vegan 包中 metaMDS() 执行,排序轴设定为 2)
nmds <- metaMDS(phylum, distance = 'bray', k = 2)
#被动添加环境变量的过程和上述 tb-PCA 类似,简要演示
#环境变量与非约束轴的多元回归,999 次置换检验
nmds_ef <- envfit(nmds~., data = env, perm = 999, choices = c(1,2), display = 'sites')
#p 值校正
nmds_ef$vectors$pvals <- p.adjust(nmds_ef$vectors$pvals, method = 'bonferroni')
#NMDS 作图,仅展示样方排序,不展示物种排序
plot(nmds, choices = c(1, 2), type = 'n')
points(nmds, choices = c(1, 2), pch = 20, col = c(rep('red', 9), rep('orange', 9), rep('green3', 9)))
#将 p.adj < 0.05 的结果投影至 NMDS
plot(nmds_ef, choices = c(1, 2), p.max = 0.05)
ordisurf()的非线性拟合
如果想观测排序轴和环境变量之间的非线性关系(而非使用多元回归),或者不期望将环境变量直接与排序轴相关联,vegan包中的ordisurf()函数可以作为替代。ordisurf()使用GAM模型(generalized additive model)实现环境变量与非约束轴的非线性回归(实际是做趋势面),并可将环境变量的拟合结果添加到现有的排序图中。
该方法对于PCA、CA、PCoA、NMDS均适用,继续以上述tb-PCA结果为例。
##将环境变量以非线性曲面的形式投影至排序空间中,可借此探索排序样本沿环境变量的梯度分布
#以两组环境变量(DOC、AK)为例
plot(tbpca, choices = c(1, 2), scaling = 1, display = c('wa'), main = 'DOC + AK')
gam1 <- ordisurf(tbpca, env[, 'DOC'], add = TRUE, col = 'red3')
gam2 <- ordisurf(tbpca, env[, 'AK'], add = TRUE, col = 'green3')
#可以再把上述 envfit() 的线性拟合结果添进去,只显示环境变量“DOC”和“AK”
select <- which(rownames(tbpca_ef_adj$vectors$arrows) %in% c('DOC', 'AK'))
tbpca_ef_adj_select <- tbpca_ef_adj
tbpca_ef_adj_select$vectors$arrows <- tbpca_ef_adj_select$vectors$arrows[select, ]
tbpca_ef_adj_select$vectors$r <- tbpca_ef_adj_select$vectors$r[select]
tbpca_ef_adj_select$vectors$pvals <- tbpca_ef_adj_select$vectors$pvals[select]
plot(tbpca_ef_adj_select, choices = c(1, 2), add = TRUE, col = c('red3', 'green3'))
#作图结果默认绘制,以下简要查看回归详情
gam1
summary(gam1)
命令中使用参数add = TRUE,可以将本次的结果添加在上一个图中,合并展示。
结果如下所示。第一张图展示作图结果,ordisurf()计算时默认绘制,各样方所处的环境梯度(曲面)在排序图中清晰可见(并未改变原排序图中样方的排列位置);第二张图展示的GAM回归的详细统计信息,包含回归的显著性p值、拟合R2,残差信息等,用以判断环境变量与非约束轴的拟合优度,多环境变量时还可相互之间比较重要程度。
常见问题与说明
关于在NMDS中拟合解释变量的可行性
将外部环境变量投影到物种变量的NMDS排序图上,这种做法也是一直在被讨论是否是合理/可能/允许的。vegan包的作者之一Jari Oksanen博士在vegan常见问题列表中给出了对这一问题的看法(问题解答2.7),指出这种做法是完全可行的,同时也对NMDS相关的一些误解作出了回应。
vegan常见问题列表:http://vegan.r-forge.r-project.org/FAQ-vegan.html
环境变量向量(箭头)的排序坐标(顶点坐标)是否能够同比例缩放
当然是可以的。在生态学统计的排序分析中(通常为了更好地用于反映生态学现象,解释生物意义,而可能会与严格的数学意义有出入),解释变量(通常为环境变量)向量(箭头)自身的长度相对于样方(点)来讲没有特殊的意义(二者的关系通过样方点在环境向量上的垂直投影的位置来反映),各环境变量的相对重要性也是通过各向量所展示的相对长度来体现的。若使用envfit()计算的原始坐标作图时,发现箭头长度过长或过短影响对排序图内容的解读时,可尝试对所有箭头的长度统一调整,同比例地放大或缩小一定倍数后最终使用合适的箭头长度作图。
对于vegan打包好的plot()相关的函数(一系列作图命令可使用?plot.cca查看),可以使用arrow.mul参数设置环境变量向量(箭头)的长度展示大小。
详细信息同样参见Jari Oksanen博士在vegan常见问题列表中的阐述(问题解答2.16)。
vegan常见问题列表:http://vegan.r-forge.r-project.org/FAQ-vegan.html
关于在约束轴中使用envfit()拟合环境变量
若我们同时使用物种变量矩阵(作为响应变量)与环境变量矩阵(作为解释变量),构建约束模型(例如RDA或CCA等),并使用envfit()将解释变量投影到约束轴中同时检验它们的重要性,这种做法是完全不可以的。
下面阐述这种做法不可行的原因,参考自David Zelený博士的阐述:https://www.davidzeleny.net/anadat-r/doku.php/en:confusions。
首先由于约束模型在计算时即已明确地探索了响应变量矩阵和解释变量矩阵的关系,本身即为物种变量与环境变量进行多元线性回归的拟合值的排序,因此环境变量信息在一开始就已经包含在约束模型内了。若我们再使用envfit()将相同的环境变量投影至约束轴,那么结果将和当前的是一致的(多此一举)。
如下示例,以RDA为例。
#使用 Hellinger 转化后的物种数据执行RDA(此时即tb-RDA),以 4 个环境变量数据为例
rda_tb <- rda(phylum_hel~., env[ ,c('DOC', 'AP', 'AK', 'NH4')], scale = FALSE)
#拟合环境变量(与样方得分)
rda_ef_site <- envfit(rda_tb~., env[ ,c('DOC', 'AP', 'AK', 'NH4')], perm = 999, choices = c(1,2), display = 'sites')
rda_ef_site$vectors$pvals <- p.adjust(rda_ef_site$vectors$pvals, method = 'bonferroni')
#拟合环境变量(与物种得分)
rda_ef_lc <- envfit(rda_tb~., env[ ,c('DOC', 'AP', 'AK', 'NH4')], perm = 999, choices = c(1,2), display = 'lc')
rda_ef_lc$vectors$pvals <- p.adjust(rda_ef_lc$vectors$pvals, method = 'bonferroni')
#作图观察
plot(rda_tb, display = c('wa', 'cn'), col = 'blue') #RDA
plot(rda_ef_site, col = 'red') #与 RDA 样方得分的拟合
plot(rda_ef_lc, col = 'green3') #与 RDA 物种得分的拟合
下图左图为RDA排序结果(展示前两轴,I型标尺);右图为envfit()拟合后的结果,在左图的基础上直接添加,发现绿色的向量与蓝色的向量完全重叠(遮盖住了)。根据RDA的算法(简略描述:http://blog.sciencenet.cn/blog-3406804-1182485.html),RDA在最初的多元回归中即首先计算了物种变量与环境变量的线性关系,因此很容易推测,若我们再使用相同的环境变量再以多元回归的方式与物种得分进行拟合,那么结果将是一摸一样的。同时由此也可知,在约束模型中拟合样方得分的做法更是不可取的(非约束轴中,拟合样方得分或物种得分均可以,视情况考虑使用哪种)。
既然使用相同的环境变量多此一举,那么我们使用未包含在约束模型中的环境变量,将它们与约束轴进行多元回归拟合可以吗?这样就更不可行了。因为约束排序结果一旦生成,排序空间内的样方以及物种信息就已经被当前所使用的环境变量“约束”了,只能通过当前给定的环境变量来解释,任何额外添加的环境变量(约束模型本身不包括的)都是无意义的。这种情况下,若想再结合补充变量,还需将补充环境变量与先前给定的环境变量合并后,重新构建约束模型。
其次,就是关于显著性的问题。由上述可知,使用envfit()将原有的环境变量(解释变量)重新拟合至RDA轴上,结果将和已有的RDA一摸一样。既然完全一致的结果,那么即可推断,envfit()所得环境变量拟合结果的显著性将会非常理想。那么,这个显著性能否说明什么问题吗?不,它没有任何意义。
以下使用3个示例,以展示为什么不能使用envfit()检验约束轴中解释变量的显著性。这里生成了一组随机变量,将其视为“环境变量”。很显然这组随机的“环境变量”没有任何生态意义,它与物种多度没有任何关系,因此我们大可预先推测检验结果是不显著的。首先计算了tb-PCA,并使用envfit()将随机变量拟合到PCA排序轴,结果不显著,如预期的那样;然后使用这组随机变量计算tb-RDA,并检验RDA轴的显著性,结果不显著,如预期的那样;最后使用envfit()将随机变量拟合到RDA排序轴,然而发现结果非常显著!因此我们可知,使用envfit()将环境变量拟合到基于相同环境变量的
示例参考文献:
沼气池的污泥与猪粪协同发酵与微生物活性关联探究。有无猪粪便添加的污泥发酵物,按时间点采样,共58份样本进行16S V4区测序分析。如图1,开展常规的物种与环境因子关联分析,并基于Bray-curtis距离的PCoA分布,进行了Envfit检验,如下图图,使用文字进行了对应描述,揭示了NH4等环境因子与群落结构关联的显著性 。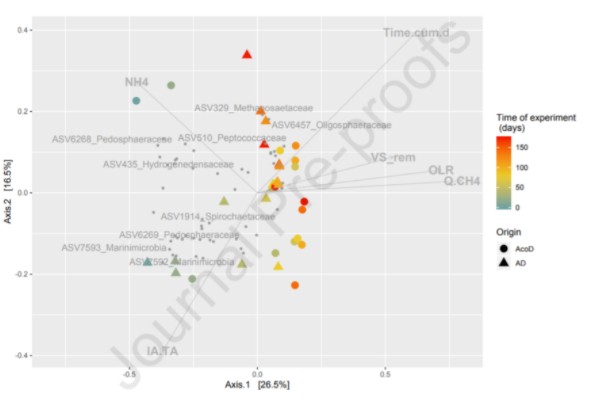

https://www.sciencedirect.com/science/article/abs/pii/S0960852419316724
- 发表于 2020-11-22 20:03
- 阅读 ( 14966 )
- 分类:宏基因组
