进化树构建方法及原理
进化树的构建
(1)数据准备
在进行系统发育分析时需要通过构建系统发育树来描述不同物种或者基因之间的进化关系,通过同源DNA的核苷酸序列或者同源蛋白质分子的氨基酸序列可以实现构建进化树的构建。
(3)序列比对
为了保证序列的同源性和所得系统发育关系的可靠性,需要对原始序列进行比对和校正。自动比对序列的软件包括Clustalw 、MAFFT、MUSCLE等。
(4)保守区用于构建进化树
保守区选择是系统发育分析过程中一个重要的步骤。分析时可以选择保守位点,也可以选择基因全长序列,但是当序列差异大时,建议保留保守序列用于进化树构建。常用的保留序列保守区的软件有Gblock、MEME等。
进化树构建方法的选择
| ML,Maximum likelihood | 最大似然法 |
| NJ,Neighbor-Joining | 邻接法 |
| MP,Maximum parsimony | 最大简约法 |
| ME,Minimum Evolution | 最小进化法 |
| Bayesian | 贝叶斯推断 |
| UPGMA | 不常用 |
第一种:UPGMA法(非加权配对算术平均法)
前提条件:在进化过程中,每一代发生趋异的次数相同,即碱基或氨基酸的替换速率是均等且恒等的。
UPGMA法计算原理和过程:
①以已求得的距离系数,所有比较的分类单元的成对距离构成一个t×t方阵,即建立一个距离矩阵M。
②对于一个给定的距离矩阵,寻求最小距离值Dpq。
③定义类群p和q之间的分支深度Lpq=Dpq/2。
④若p和q是最后一个类群,侧聚类过程完成,否侧合并p和q成一个新类群r。
⑤定义并计算新类群r到其他各类群i(i≠p和q)的距离Dir=(Dpi+Dqi)/2。
⑥回到第一步,在矩阵中消除p和q,加入新类群r,矩阵减少一阶,重复进行直至达到最后归群。
UPGMA法比较直观和简单,运算速度快,应用很广。它的缺点在于当分子进化速率较大时,在建树过程会引入系统误差。
第二种:邻接法NJ法(neighbor joining method)
是一种推论叠加树的方法。在概念上与UPGMA法相同,但是有四点区别
a. NJ法不要求距离符合超度量特性,但要求数据应非常接近或符合叠加性条件,即该方法要求对距离进行校正。
b. 邻接法在成聚过程中连接的是分类单元之间的节点(node),而不是分类单元本身。
c. NJ法中原始距离数据用于估算系统树上所有端结分类单元之间的距离矩阵,校正后的距离用于确定节点之间的连接顺序。
d. 在重建系统发育树时,NJ法取消了UPGMA法所做的假定,认为在此进化分支上,发生趋异的次数可以不同。
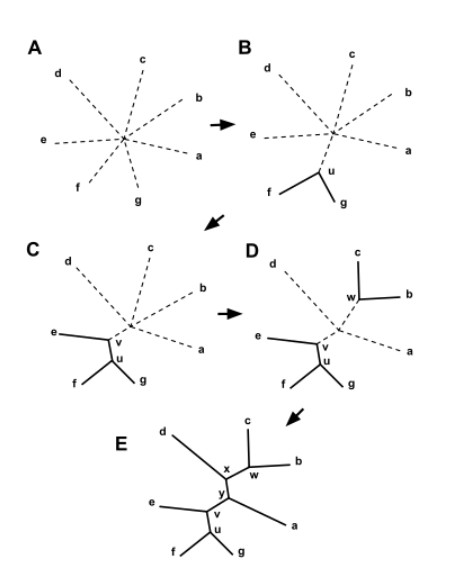
① 对于给定距离矩阵中的每一端结i,用下式计算与其它分类单元之间的净趋异量(Ri) (t:矩阵中的分类单元数)
② 建立一个速率校正距离矩阵M,其元素由下式确定:
③ 定义一个新节点u,u的三个分支分别与节点i,j和树的其余部分相连,并且Dij为矩阵中距离最小者,u到节点i和j的分支长度定义为
④ 定义u到树的其它节点k(k≠i和j外的所有节点)的距离:
⑤ 从距离矩阵中删除i和j的距离,矩阵减少一阶。
⑥ 如果矩阵仍然多于两个的节点,重复第①-⑤步,否测除最外两个节点的分支长度来确定外,树上其余节点都确定,最后是剩余的2个的分支长度Sy=Dij
第三种:最大简约法(Maximum Parsimony Method)
依据 基于奥卡姆(Ockham)哲学原则,这个原则认为:解释一个过程的最好理论是所需假设数目最少的那一个。
方法 计算所有可能的拓扑结构 ,计算出所需替代数最小的那个拓扑结构,作为最优树。
特点 用于分析如插入、缺失等序列。在分析序列上存在较多的回复突变或平行突变,而被检验的序列位点数又比较少的时候,最大简约法可能会给出一个不合理的或者错误的进化树推导结果。
第四种:最大似然法
依据:这个方法最早是遗传学家以及统计学家罗纳德·费雪爵士在 1912 年至1922 年间开始使用的 。基本思想是:当从模型总体随机抽取n组样本观测值后,最合理的参数估计量应该使得从模型中抽取该n组样本观测值的概率最大,而不是像最小二乘估计法旨在得到使得模型能最好地拟合样本数据的参数估计量。
方法:选取一个特定的替代模型来分析给定的一组序列数据,使得获得的每一个拓扑结构的似然率都为最大值,然后再挑出其中似然率最大的拓扑结构作为最优树(所以分析时间比较长)
特点:最大似然法具有很好的统计学理论基础,是一个比较成熟的统计学方法。选择合理的模型后,最大似然法可以推导出一个效果很好的进化树结果。但是对于相似度很低的序列,NJ往往出现Long-branch attraction(LBA,长枝吸引现象),有时严重干扰进化树的构建。
小结
比较以上几种主要的构树方法,一般情况下,若有合适的分子进化模型可供选择,用最大似然法构树获得的结果较好;对于近缘物种序列,通常情况下使用最大简约法;而对于远缘物种序列,一般使用邻接法或最大似然法。对于相似度很低的序列,邻接法往往出现长枝吸引(branch attraction)现象,有时严重干扰进化树的构建。对于各种方法重建进化树的准确性,Hall (2005)认为贝叶斯法最好,其次是最大似然法,然后是最大简约法。
Hall BG. Comparison of the accuracies of several phylogenetic methods using protein and DNA sequences. Mol Biol Evol. 2005 Mar;22(3):792-802. doi: 10.1093/molbev/msi066. Epub 2004 Dec 8. Erratum in: Mol Biol Evol. 2005 Apr;22(4):1160. PMID: 15590907.
- 发表于 2020-09-28 15:50
- 阅读 ( 16069 )
- 分类:遗传进化
