检查了示例文件,这地方的基因型有些特殊,不标准,导致beagle 报错: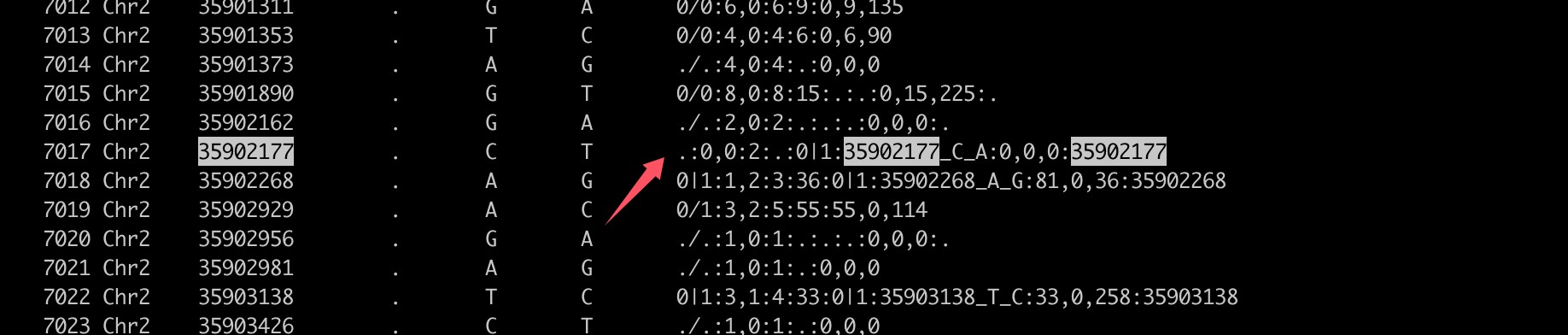
运行下面的代码修改一下
zcat ../00.filter/clean.vcf.gz |sed 's#\t.:#\t./.:#g' |gzip - > clean.vcf.gz
新文件输入运行beagle
beagle -Xmx4g -Djava.io.tmpdir=./ gt=clean.vcf.gz out=all.impute impute=true window=10 nthreads=2
检查了示例文件,这地方的基因型有些特殊,不标准,导致beagle 报错:
运行下面的代码修改一下
zcat ../00.filter/clean.vcf.gz |sed 's#\t.:#\t./.:#g' |gzip - > clean.vcf.gz
新文件输入运行beagle
beagle -Xmx4g -Djava.io.tmpdir=./ gt=clean.vcf.gz out=all.impute impute=true window=10 nthreads=2
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!

vcftools --gzvcf all.varFilter.vcf.gz --recode --recode-INFO-all --stdout --maf 0.05 --max-missing 0.9 --minDP 2 --maxDP 1000 \
--minQ 30 --minGQ 0 --min-alleles 2 --max-alleles 2 --remove-indels |gzip - > clean.vcf.gz
这条命令的--min-alleles 2 --max-alleles 2可以把多等位位点过滤掉吧?或者是其他命令能过滤掉?