微生物16S分析时候,两端测序数据拼接
老师您好!我在进行16S微生物组数据双端合并的时候,执行这项代码: qiime vsearch merge-pairs \
--i-demultiplexed-seqs primer-trimmed-demux.qza \
--p-threads 7 \
--o-merged-sequences demux-joined.qza后,查看合并结果,发现合并前后数据少了特别多。例如:合并前正反向序列均为3万多,合并后就剩下4000多了,想问下是什么原因造成的?还能用于后续的分析吗?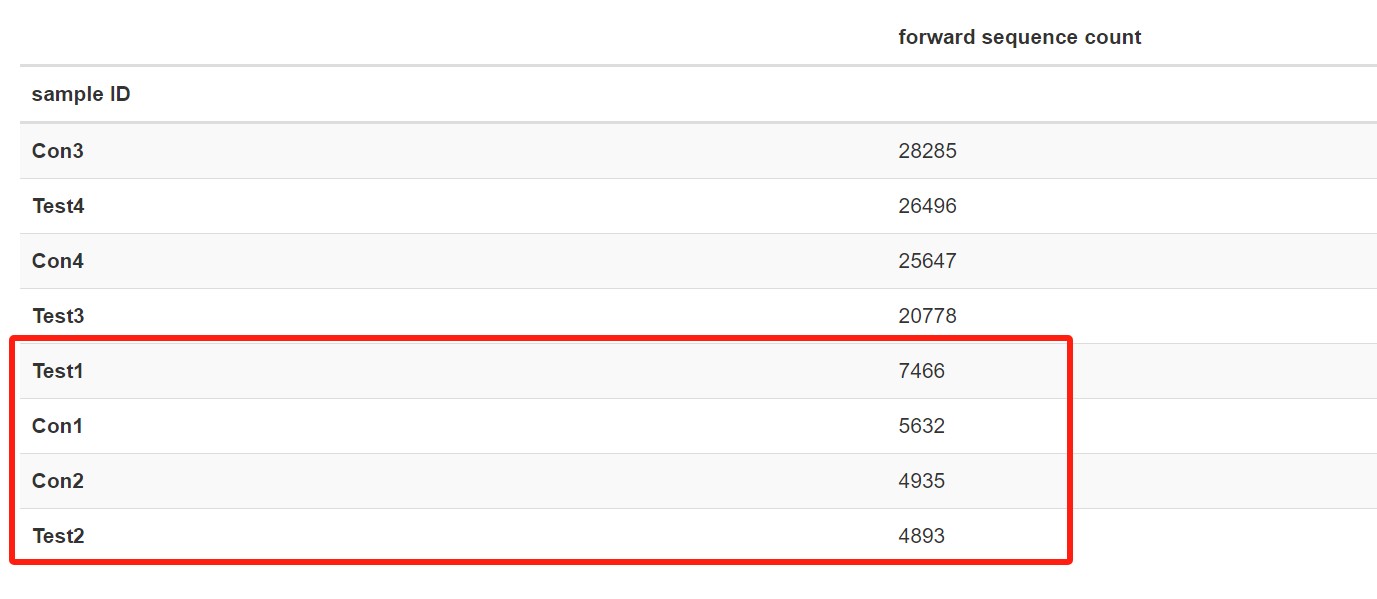
最佳答案 2024-01-29 15:13
的确差异挺大的;;
看看这几个样本测序配置文件有没有搞错;合并双端数据看看read1 read2 overlap参数放低看看:
Usage: qiime vsearch merge-pairs [OPTIONS]
Merge paired-end sequence reads using vsearch's merge_pairs function. See
the vsearch documentation for details on how paired-end merging is
performed, and for more information on the parameters to this method.
Inputs:
--i-demultiplexed-seqs ARTIFACT SampleData[PairedEndSequencesWithQuality]
The demultiplexed paired-end sequences to be merged.
[required]
Parameters:
--p-truncqual INTEGER Truncate sequences at the first base with the
Range(0, None) specified quality score value or lower. [optional]
--p-minlen INTEGER Sequences shorter than minlen after truncation are
Range(0, None) discarded. [default: 1]
--p-maxns INTEGER Sequences with more than maxns N characters are
Range(0, None) discarded. [optional]
--p-allowmergestagger / --p-no-allowmergestagger
Allow merging of staggered read pairs.
[default: False]
--p-minovlen INTEGER Minimum length of the area of overlap between reads
Range(0, None) during merging. [default: 10]
--p-maxdiffs INTEGER Maximum number of mismatches in the area of overlap
Range(0, None) during merging. [default: 10]
--p-minmergelen INTEGER
Range(0, None) Minimum length of the merged read to be retained.
[optional]
--p-maxmergelen INTEGER
Range(0, None) Maximum length of the merged read to be retained.
[optional]
--p-maxee NUMBER Maximum number of expected errors in the merged read
Range(0.0, None) to be retained. [optional]
--p-threads INTEGER Range(0, 8, inclusive_end=True)
The number of threads to use for computation. Does
not scale much past 4 threads. [default: 1]
Outputs:
--o-merged-sequences ARTIFACT SampleData[JoinedSequencesWithQuality]
The merged sequences. [required]
Miscellaneous:
--output-dir PATH Output unspecified results to a directory
--verbose / --quiet Display verbose output to stdout and/or stderr
during execution of this action. Or silence output if
execution is successful (silence is golden).
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
There was a problem with the command:
再不行你找找你的测序公司,测序质量是不是有问题;
有质量问题,可以删除这些样本;
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!
