这个报错没遇到过,新版的GATK不推荐用这个方法了,用:GenomicsDBImport 导入数据库,再 GenotypeGVCFs
具体可以看课程:https://bdtcd.xetslk.com/s/1VQOjQ
老师们好,我最近在合并300份样品的gVCF,但是遇到了如题目所示的问题。
我的重测序物种染色体较长,我将每条染色体按350000000bp+剩余长度进行了分段,并分染色体进行gVCF合并。目前,只有一条染色体出现如题所示的问题(长度为350000000bp),其余染色体均正常运行(包括几条350000000bp长度的染色体)。
我的GATK版本是4.3.0.0,运行脚本如下 gatk CombineGVCFs --java-options "-Xms100g -Xmx100g -XX:ParallelGCThreads=4" --use-jdk-inflater true --use-jdk-deflater true --reference /path/to/part.fasta $(for i in {001..300}; do echo "--variant /path/to/part_gvcf/${i}.g.vcf.gz " ;done) --intervals chr2_part1 --output /path/to/part_gvcf/chr2_part1.g.vcf.gz --tmp-dir /path/to/temp,其中“--use-jdk-inflater true --use-jdk-deflater true”是我查看了GATK官网后加上的,但是依然报错。
报错信息如下:
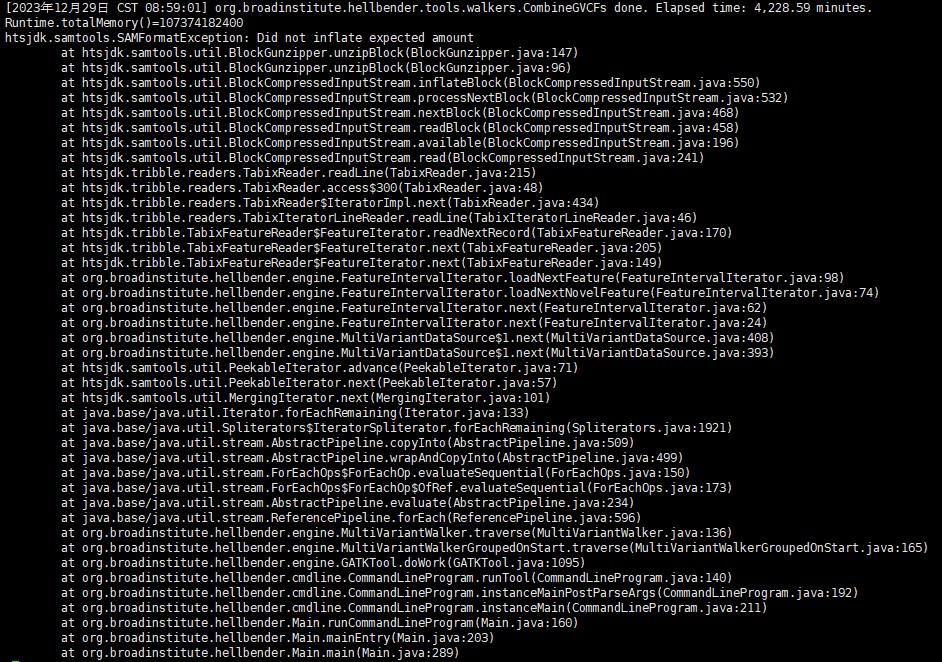 麻烦各位老师帮我解决一下,非常感谢!
麻烦各位老师帮我解决一下,非常感谢!
