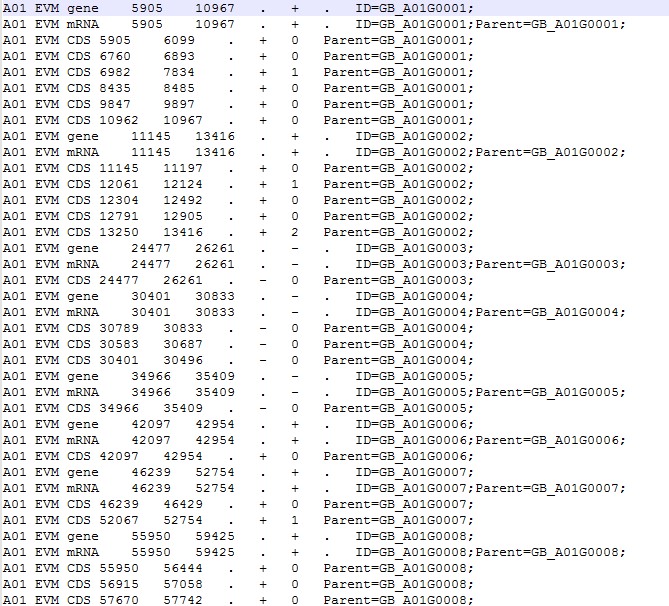
 这是我的ID
这是我的ID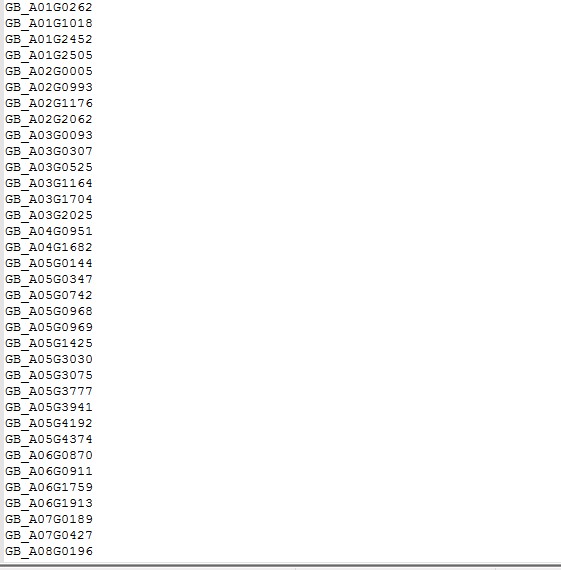
脚本用错了吧,你可以把perl脚本贴一下我看看问题
2 个回答

use Getopt::Long;
my %opts;
use Data::Dumper;
GetOptions( \%opts, "in1=s", "in2=s", "out=s", "h" );
if ( !defined( $opts{in1} )
|| !defined( $opts{in2} )
|| !defined( $opts{out} )
|| defined( $opts{h} ) )
{
&USAGE;
}
open( IN1, "$opts{in1}" ) || die "open $opts{in1} failed\n";
open( IN2, "$opts{in2}" ) || die "open $opts{in2} failed\n";
open( OUT, ">$opts{out}" ) || die "open $opts{out} failed\n";
my %gffs;
while (<IN1>) {
chomp;
next if /^#/;
my @b = split/\st/, $_;
$gffs{$b[0]} = 1;
}
#print Dumper(\%gffs);
while (<IN2>) {
chomp;
next if (/^#/);
my @a = split /\t/, $_;
next if $a[2]=~/exon/i;
if ($a[2] =~/^mRNA$/i or $a[2] =~/^transcript$/i ) {
($id1) = ($a[8] =~ m/ID=([^;]*)/);
}elsif ( $a[2] =~/^CDS$/i or $a[2] =~/utr/i ) {
($id1) = ($a[8] =~ m/Parent=([^;]*)/);
}else{
next;
}
if ( exists $gffs{$id1} ) {
print OUT "$_\n";
}
}
close OUT;
close IN1;
close IN2;
sub USAGE {
print "usage: perl $0 -in1 mRNA_id.txt -in2 genome.gff3 -out gene_location.txt ";
exit;
}
老师,这是全部脚本内容