转录组差异基因为什么要进行聚类分析?
聚类分析用于判断差异基因在不同实验条件下的表达模式,将表达模式相同或相近的基因聚集成类,进而识别未知基因的功能或已知基因的未知功能,这些同类基因可能具有相似的功能,共同参...
- 0
- 2
- landy
- 发布于 2018-06-28 16:28
- 阅读 ( 18108 )
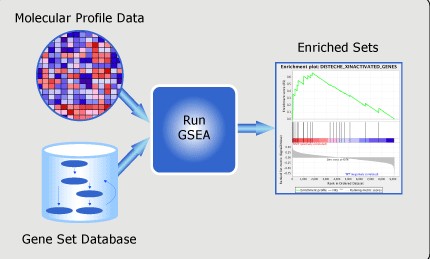
如何将GEO芯片注释文件整理成GSEA能使用的芯片注释文件?
将GEO数据库中的注释文件整理成GSEA软件可以使用的芯片注释文件
- 0
- 2
- landy
- 发布于 2018-06-22 15:01
- 阅读 ( 10343 )
Excel数据分析技巧之双因素方差分析
>>方差分析之双因素方差分析 小编在节前做过一期使用excel表进行单因素方差分析的推送,反响不错,小编也颇受鼓舞!但是,在许多情况下,单因素方差分析已经不能满足实际分析的需要。比...
- 1
- 2
- 生信老顽童
- 发布于 2018-06-20 10:29
- 阅读 ( 7354 )
TCGAbiolinks 错误 Error in value[[3L]](cond) : GDC server down, try to use this package later
TCGAbiolinks 错误 Error in value[[3L]](cond) : GDC server down, try to use this package later 排查
- 0
- 2
- microRNA
- 发布于 2018-06-20 09:47
- 阅读 ( 15924 )
aggregate对数据分组处理
利用aggregate对数据进行分组处理,包括分组求和,分组取均值,最大值,中位数等等
- 0
- 2
- Daitoue
- 发布于 2018-05-31 15:52
- 阅读 ( 9985 )
R语言计算平均值,中位数
Mean()求平均值 通过求出数据集的和再除以求和数的总量得到平均值 函数mean()用于在R语言中计算平均值。语法 用于计算R中的平均值的基本语法是 - mean(x, trim = 0, na.rm = FALSE, ...)...
- 1
- 2
- 安生水
- 发布于 2018-05-24 15:53
- 阅读 ( 15124 )
linux下替换文件中的换行符
之前在linux系统执行以下命令: ls file |xargs sed -i 's/\n//g' 然而却发现没有任何效果,百度之后发现 sed是按行处理文本数据的,每次处理一行数据后,都会在行尾自动添加trailing newline...
- 0
- 2
- 安生水
- 发布于 2018-05-24 10:35
- 阅读 ( 7347 )
推荐一个好用文献管理软件:CNKI E-Study——数字化学习与研究平台(1)
CNKI E-Study——数字化学习与研究平台的学习使用
- 0
- 2
- smyang2018
- 发布于 2018-05-18 19:22
- 阅读 ( 5388 )
Python虚拟环境解决不兼容问题
采用Python 虚拟环境,解决python不同版本,不同包 间不相容的问题
- 0
- 2
- microRNA
- 发布于 2018-05-18 10:11
- 阅读 ( 3619 )
pip安装python模块
python模块安装还是非常简单的,只要把pip安装好之后,一切都用一条命令搞定。 如果你使用的是从 python.org 下载的Python 2 >=2.7.9 或者Python 3 >=3.4,那么pip是已经安装了。 1....
- 1
- 2
- 安生水
- 发布于 2018-05-18 09:47
- 阅读 ( 3046 )
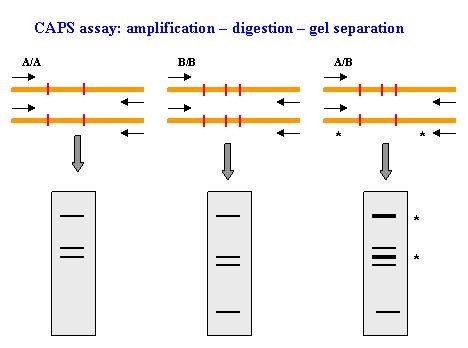
如何将SNP标记转化成可跑胶验证的CAPS标记
现如今,基于高通量测序得到的SNP信息无疑是海量的,然而SNP标记的验证却并不容易实现。其最好的验证方式是一代sanger测序,不过价格较高,而且通量低。如果SNP刚好位于酶切位点上,就可以将其转化为CAPS标记,利用跑胶的方式对其进行验证,既便宜又好用。
- 1
- 2
- 安生水
- 发布于 2018-05-18 08:46
- 阅读 ( 12888 )