泛基因家族分析结构变异分析,生成的mcoords和mdelta文件为空
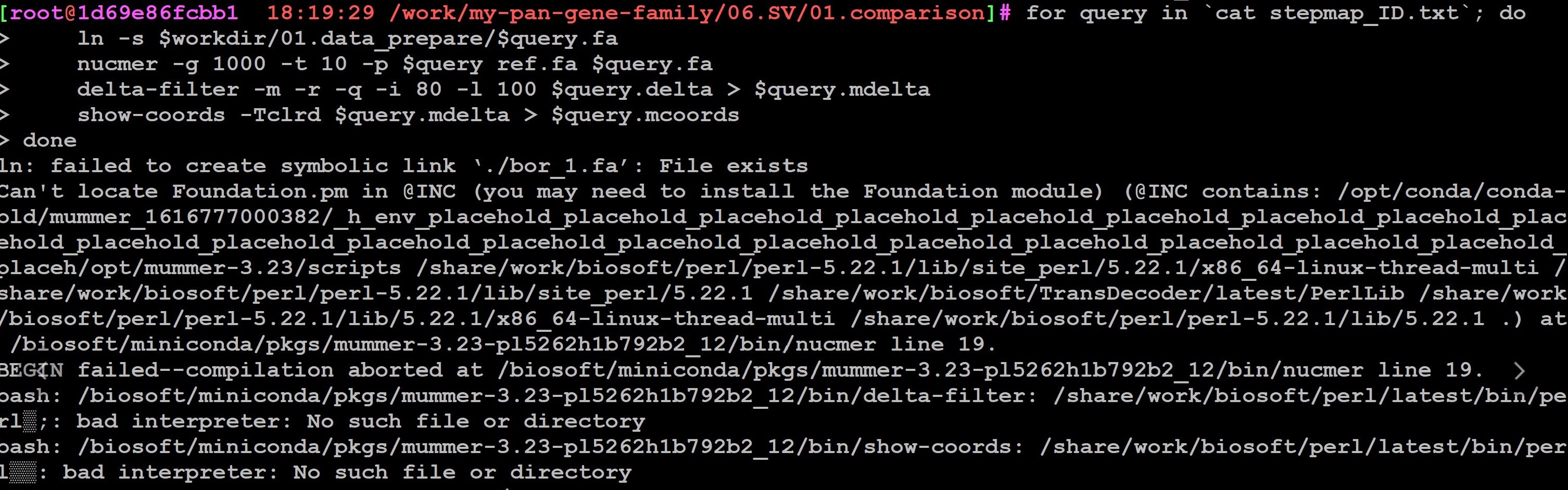 命令行是
命令行是
#numer比对
for query in `cat stepmap_ID.txt`;do
#去除未组装到染色体水平的scaffold和contig序列
#seqkit grep -r -p '^chr\d+$' $workdir/01.data_prepare/$query.fa > $query.fa
ln -s $workdir/01.data_prepare/$query.fa
# 比对,生成query.delta
nucmer -g 1000 -t 10 -p $query ref.fa $query.fa
# 多对多 比对过滤
delta-filter -m -r -q -i 80 -l 100 $query.delta > $query.mdelta
#转换成人类可读的表格格式
show-coords -Tclrd $query.mdelta > $query.mcoords
done