5 作共线性分析的时候,用水稻的bed文件,无法获得水稻的cds,是不是因为水稻的cds.fa文件内是os08t0254300-00,而不像拟南芥的at012358.1这样后缀不一样,所有无法过去它的cds
回答问题即可获得 10 经验值,回答被采纳后即可获得 10 金币。
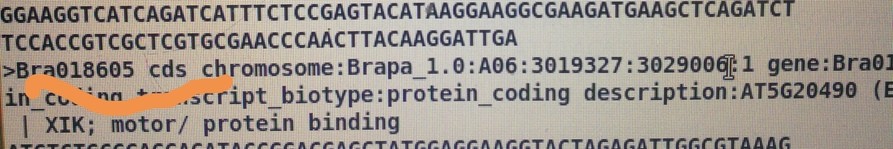
 运行这个命令的时候perl get_fa_by_id_from_bed.pl ATH.bed Arabidopsis_thaliana.TAIR10.cds.all.fa ATH.cds
运行这个命令的时候perl get_fa_by_id_from_bed.pl ATH.bed Arabidopsis_thaliana.TAIR10.cds.all.fa ATH.cds
脚本:#北京组学生物科技有限公司
#email: huangls@biomics.com.cn
die "perl $0 <idlist> <fa> <OUT>" unless ( @ARGV == 3 );
use Math::BigFloat;
use Bio::SeqIO;
use Bio::Seq;
#读入蛋白序列
$in = Bio::SeqIO->new(
-file => "$ARGV[1]",
-format => 'Fasta'
);
#输出蛋白序列:
$out = Bio::SeqIO->new(
-file => ">$ARGV[2]",
-format => 'Fasta'
);
#读取需要提取基因ID
my %keep = ();
open IN, "$ARGV[0]" or die "$!";
while (<IN>) {
chomp;
next if /^#/;
my @a = split /\t/;
$keep{"$a[3].1"}=1; ##注意提取第一个转录本
}
close(IN);
#输出想要的基因的序列
while ( my $seq = $in->next_seq() ) {
my ( $id, $sequence, $desc ) = ( $seq->id, $seq->seq, $seq->desc );
if ( exists $keep{$id} ) {
$out->write_seq($seq);
}
}
$in->close();
$out->close();
输入文件为:Oryza_sativa.IRGSP-1.0.cds.all.fa 和os.bed
结果:无法获得os的CDS序列


