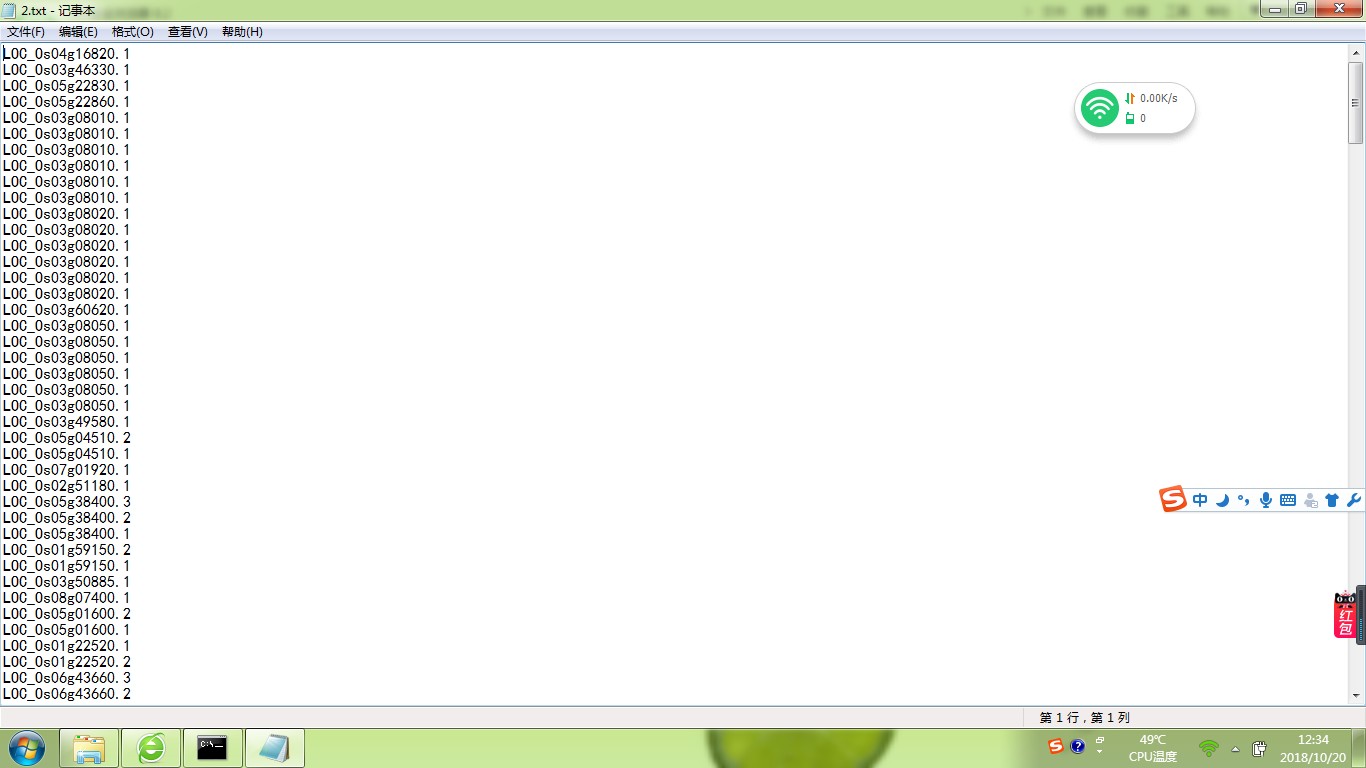
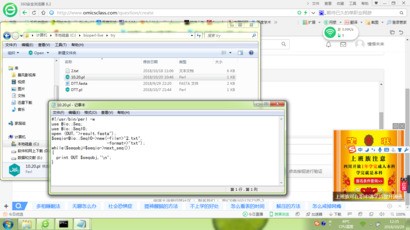
你写的脚本不对:
这里有个根据ID,提取序列的脚本,你可以参考一下:
#script www.omicsclass.com
die "perl $0 <id><fa><OUT>" unless(@ARGV==3);
use Bio::SeqIO;
use Bio::Seq;
my$in = Bio::SeqIO->new(-file => "$ARGV[1]" ,
-format => 'Fasta');
my$out = Bio::SeqIO->new(-file => ">$ARGV[2]" ,
-format => 'Fasta');
my%keep=();
open IN ,"$ARGV[0]" or die "$!";
while(<IN>){
chomp;
next if /^#/;
my@tmp=split(/\s+/);
$keep{"$tmp[1].1"}=1; #注意这里加了.1 注意转录本ID与基因ID对应
}
close(IN);
while ( my $seq = $in->next_seq() ) {
my($id,$sequence,$desc)=($seq->id,$seq->seq,$seq->desc);
if( exists $keep{$id}){
$out->write_seq($seq);
}
}
$in->close();
$out->close();
不会写脚本,可以用下工具:https://www.omicsclass.com/article/64
