20 今天python版本的MCscanX在用于比较基因组学分析时突然出了问题
更新一下细节:
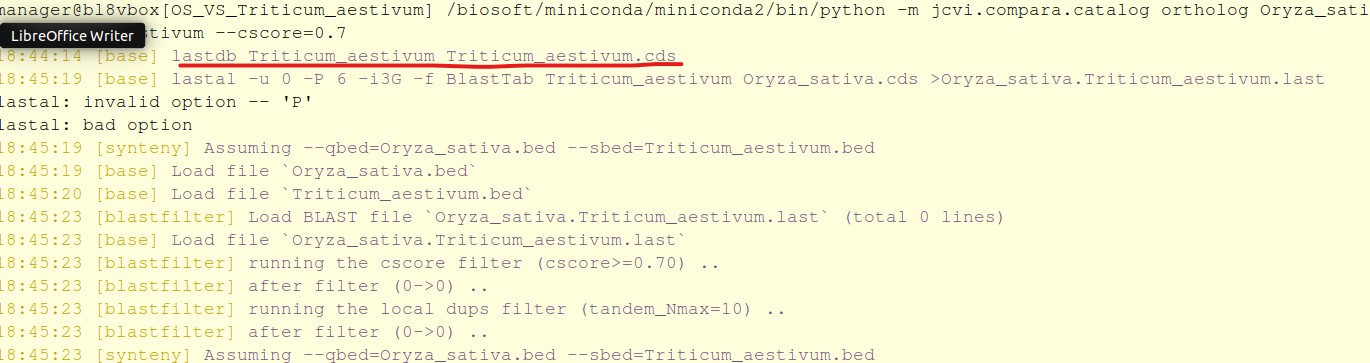
问题应该出在lastdb这一步。是不是blast出了问题?关键是我一个月之前还能用的,中间重装了一次linux系统(还是用的组学大讲堂的装机包),但是这次用就不行了。
生成的last文件为空,关键是原来可以跑成功的cds文件和bed文件这次也不行了,汗...
----------------------------------------------------------
(以下原回答)
原来是用都好好的;
但是今天用的时候用原来一样的.cds和.bed文件却一个anchor都找不到了;和原来一样的操作步骤也不行。。。
不知道为什么?
这是命令行:/biosoft/miniconda/miniconda2/bin/python -m jcvi.compara.catalog ortholog Oryza_sativa Zea_mays --cscore=0.7
这是报错:
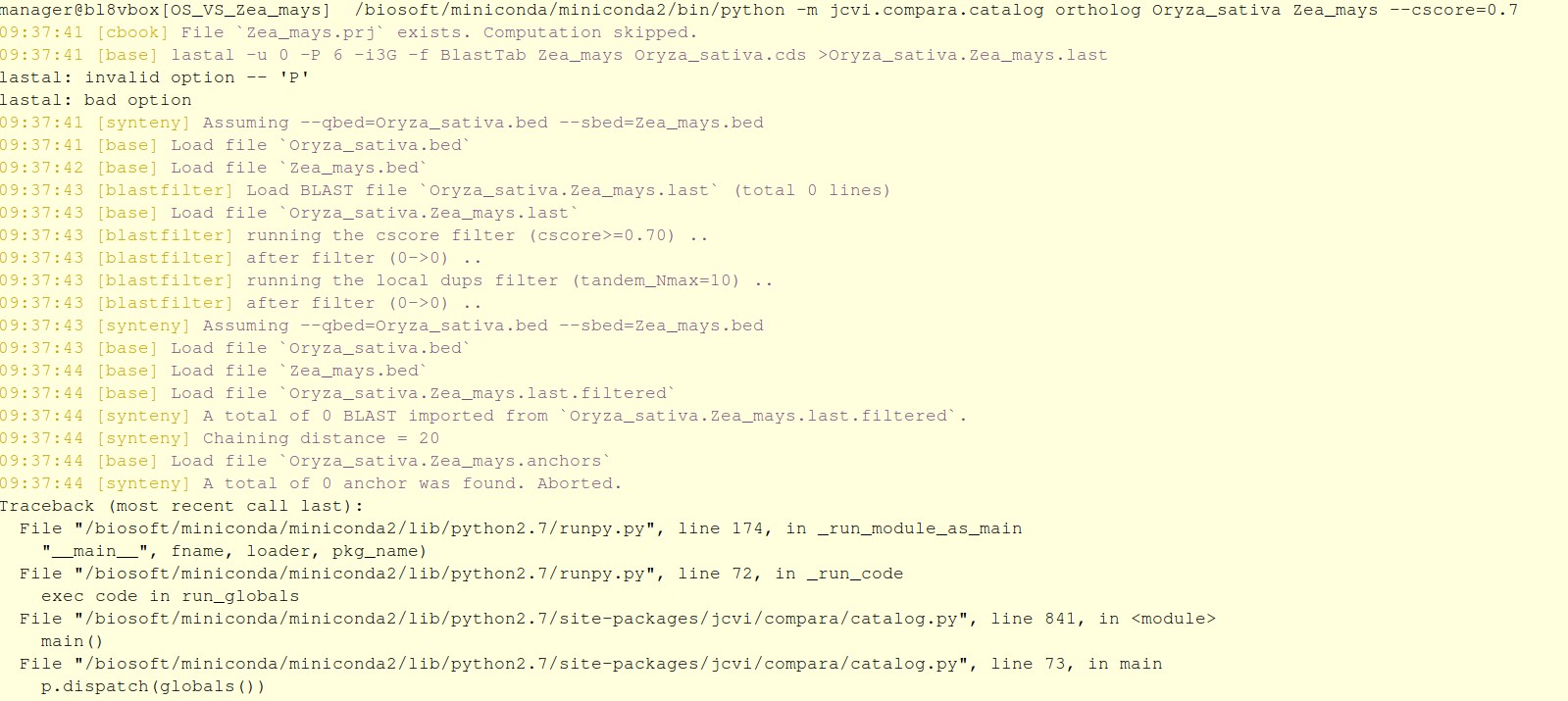
以下是输入文件:
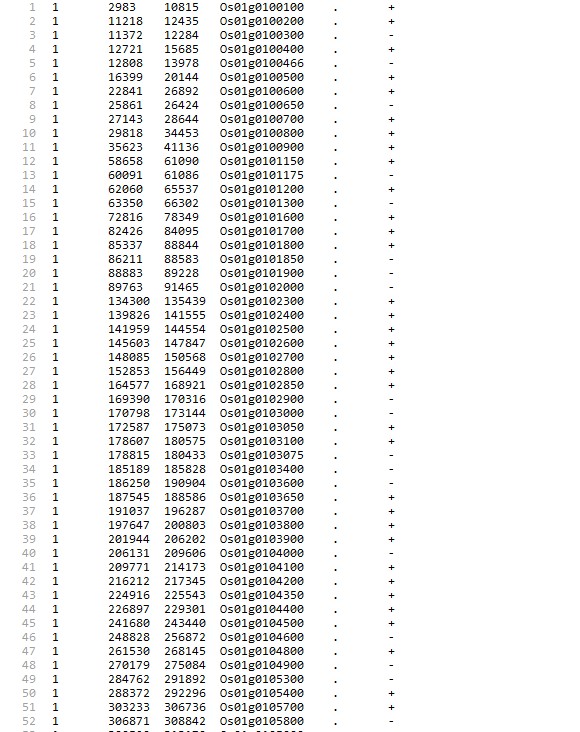
.bed:
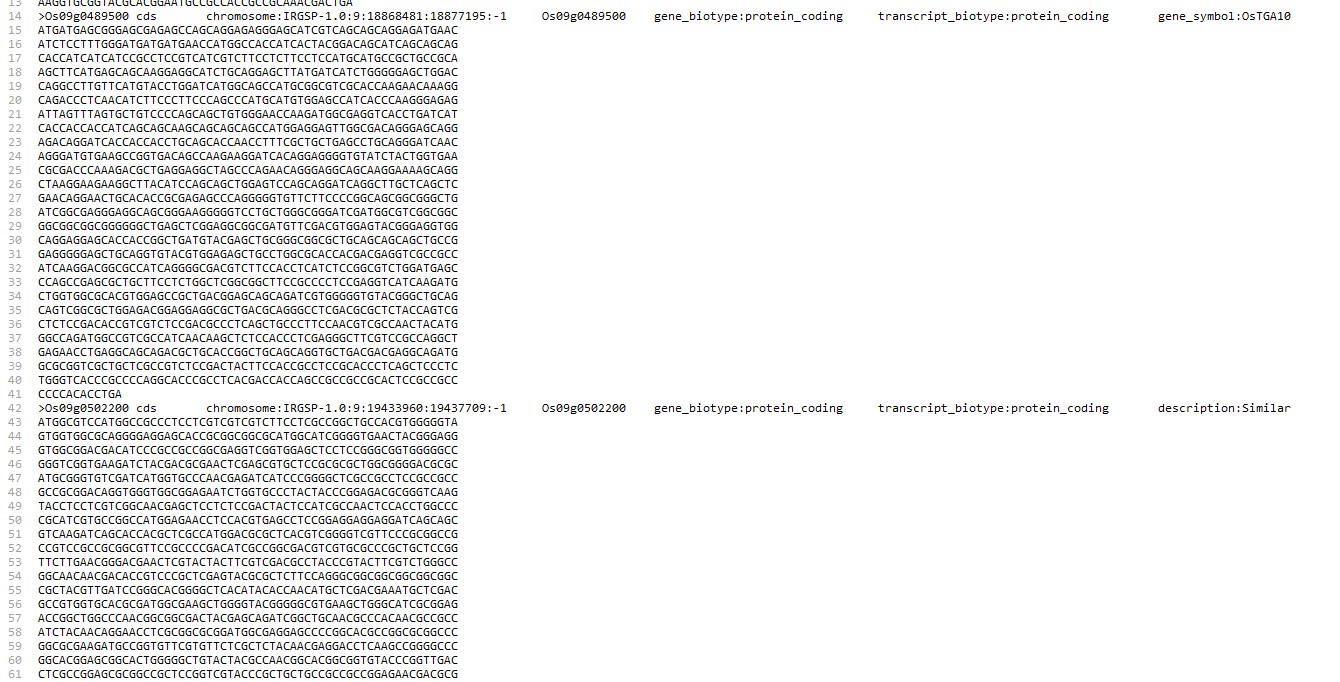
Os:
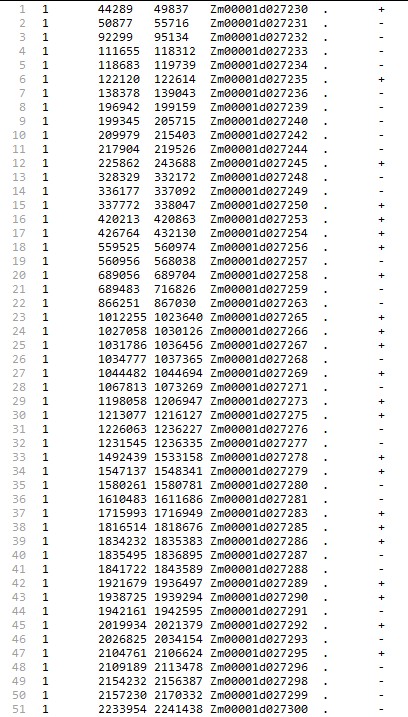
Zm:
.cds
Os: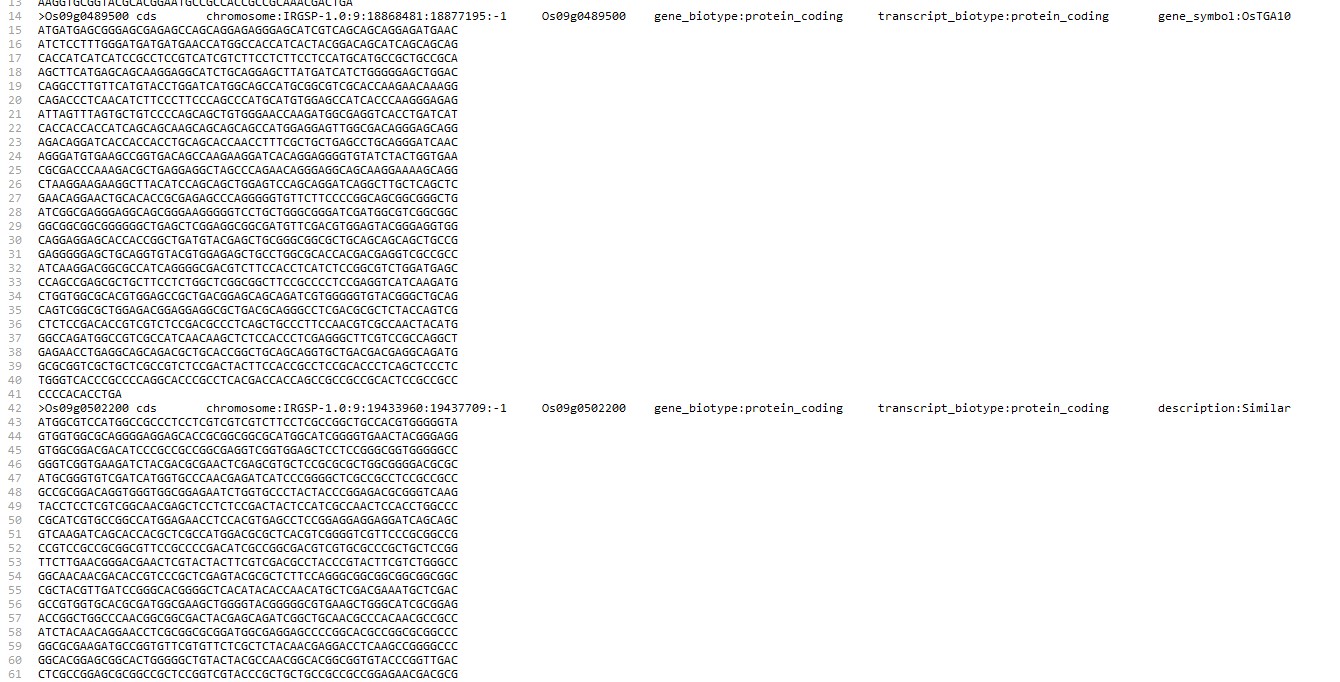
Zm:
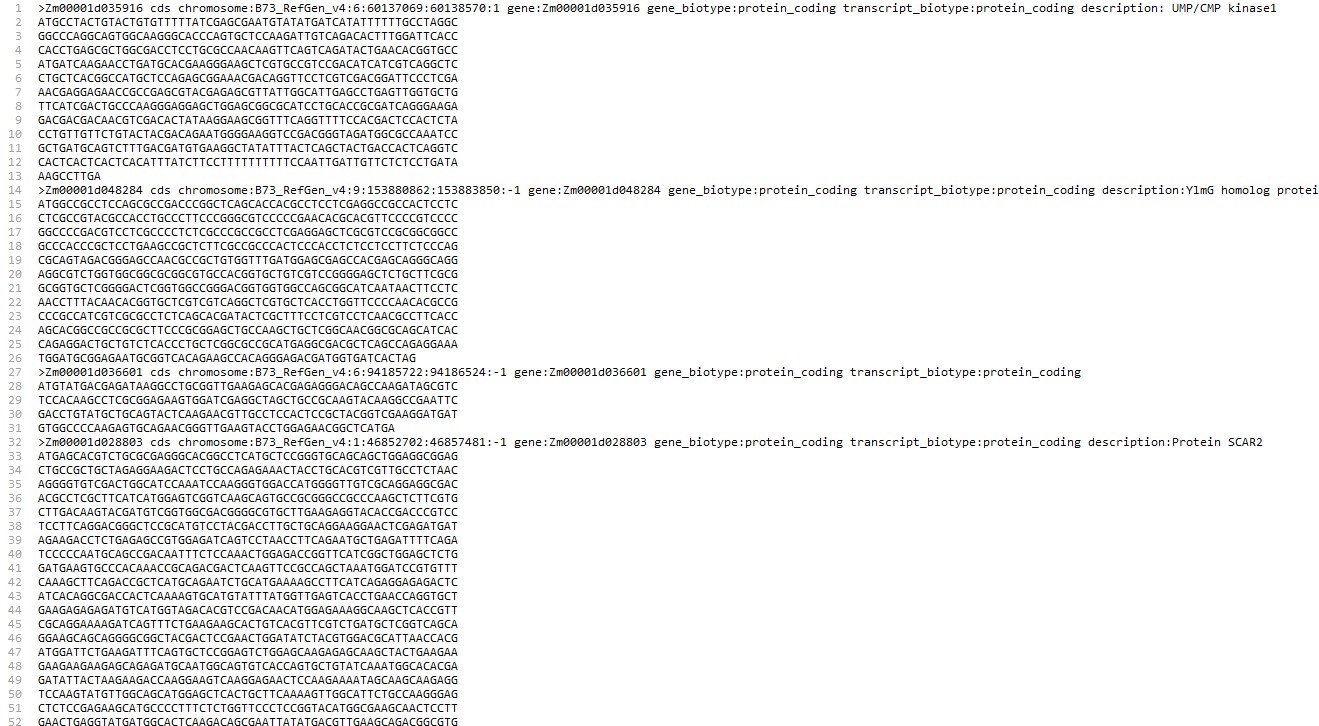
最佳答案 2020-06-23 19:17
问题已解决;
根据组学大讲堂提供的MCscanX的安装方法(https://www.omicsclass.com/article/284);重新下载了lastdb加入到环境变量中。
