IBD分析
IBD(Identity by Descent,同源片段分析)是群体遗传学中用于检测个体间共享祖先片段的分析方法。
一、IBD分析的基本概念
IBD分析是通过检测基因组中共享的、未发生重组的DNA片段,推断个体...
IBD(Identity by Descent,同源片段分析)是群体遗传学中用于检测个体间共享祖先片段的分析方法。
一、IBD分析的基本概念
IBD分析是通过检测基因组中共享的、未发生重组的DNA片段,推断个体间的亲缘关系或群体历史事件。其核心原理是:有共同祖先的个体,其基因组中会保留来自该祖先的、未被打断的长片段。
主要应用场景:
亲缘关系推断:检测亲子、同胞、堂表亲等关系,比传统方法更精细
群体遗传学研究:揭示群体混合历史、瓶颈事件、有效群体大小
医学遗传学:在病例对照研究中识别共享致病基因区域
育种学:动物/植物育种中的系谱验证
解决的核心问题:
量化个体间的遗传相似性程度
识别基因组中共享祖先片段的分布和长度
推断近期的共同祖先时间
检测群体结构或隔离
二、分析流程
1. 基因型数据的过滤
1.1 缺失率过滤,最高能接受20%的缺失
vcftools --vcf .vcf --max-missing 0.95 --maf 0.01 --recode --stdout
1.2 LD过滤
plink --vcf clean.vcf --indep-pairwise 50 10 0.2 --out tmp.ld --allow-extra-chr --set-missing-var-ids @:#
# plink --vcf clean.vcf --extract tmp.ld.prune.in --out filter.LD.tmp --recode vcf-iid --keep-allele-order --allow-extra-chr --set-missing-var-ids @:#
# plinkawk -v OFS="\t" ' {if($1!~/#/) {print "Chr"$0} else { print}} ' filter.LD.tmp.vcf > ibd.vcf
bgzip ibd.vcf
tabix ibd.vcf.gz
2. 基因型填充
java -jar /share/biosoft/beagle/beagle.jar gt=ibd.vcf.gz out=imputed_data nthreads=8
3. 运行RefinedIBD检测IBD片段
java -jar /share/biosoft/beagle/refined-ibd.jar \
gt=imputed_data.vcf.gz \
out=ibd_results \
length=1.0 \
lod=1.0 \
nthreads=8
IBD检测本质上是两两个体间某一haplotype的比较。输出结果分别为*.ibd和*.hbd;ibd表示两两个体间一致的IBD片段,hbd表示某一个体内两个haplotype完全一致即纯合子。
4. 整理绘图
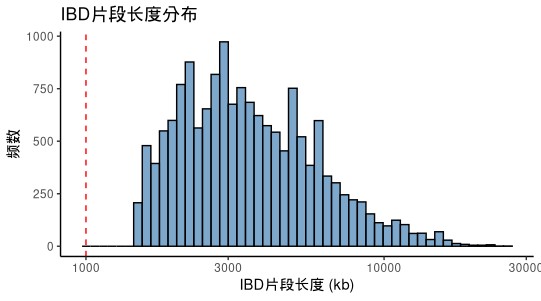
绘制直方图,建议大家可以先根据group做提取再进行绘制:
短片段多:远古共享祖先
长片段多:近期混合事件
library(ggplot2)
library(reshape2)
library(dplyr)
# ibd_segments <- read.table("ibd_results.ibd", header = FALSE,
col.names = c("sample1","hap1", "sample2","hap2", "chr", "start", "end", "lod","un"))
ibd_segments$length_bp <- ibd_segments$end - ibd_segments$start
ibd_segments$length_kb <- ibd_segments$length_bp / 1000
# ggplot(ibd_segments, aes(x = length_kb)) +
geom_histogram(bins = 50, fill = "steelblue", alpha = 0.7, color = "black") +
scale_x_log10() + # theme_classic() +
labs(x = "IBD title = "IBD geom_vline(xintercept = 1000, linetype = "dashed", color = "red")
- 发表于 2026-01-30 14:05
- 阅读 ( 874 )
- 分类:遗传进化
