带推断线的进化树
在一些群体遗传进化的文章中,会展示整合了物种分类和大量系统发育推断(灰色虚线)的进化树,接下来小编将绘图代码贴在下面,效果如下图:
1.准备数据
准备3个文件,其中两个nwk格式的树文件...
在一些群体遗传进化的文章中,会展示整合了物种分类和大量系统发育推断(灰色虚线)的进化树,接下来小编将绘图代码贴在下面,效果如下图: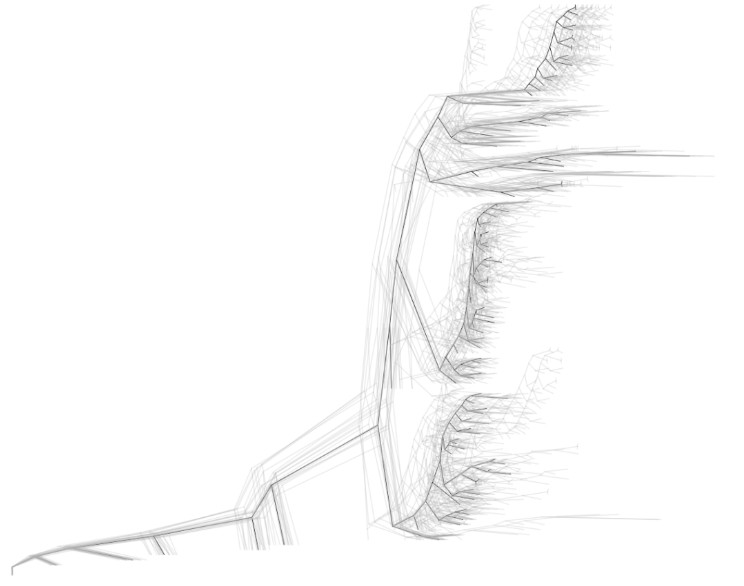
1.准备数据
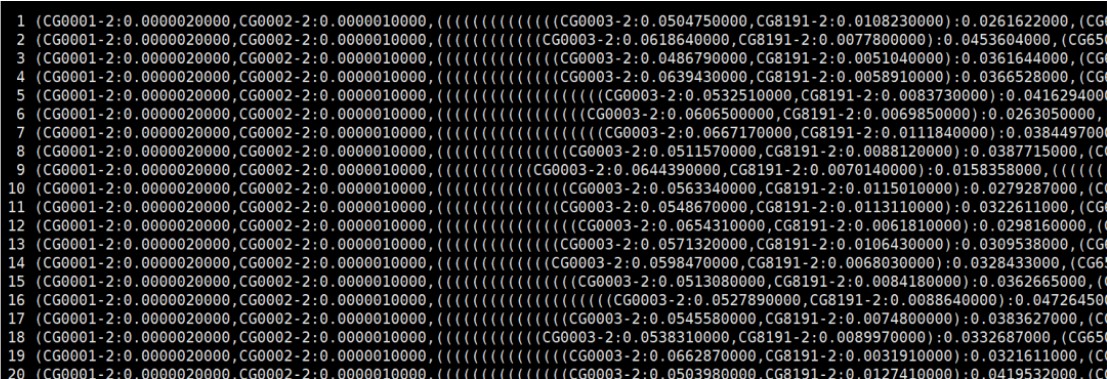
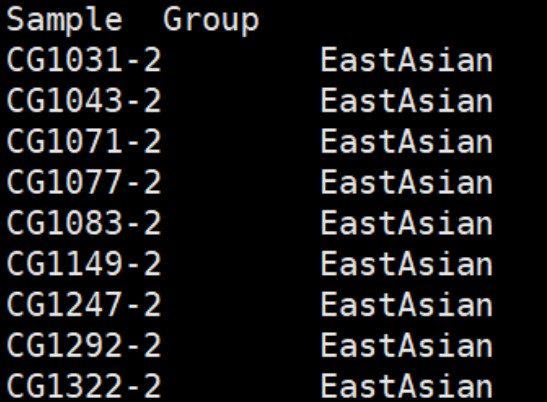
准备3个文件,其中两个nwk格式的树文件,一个为主干树(共识树,通常可以是iqtree等构树软件的最终结果),一个是自举树。另外需要准备一个样本分类的文件(group.txt),一共两列,第一列为样本名称,第二列为样本所属组别。
主干树:
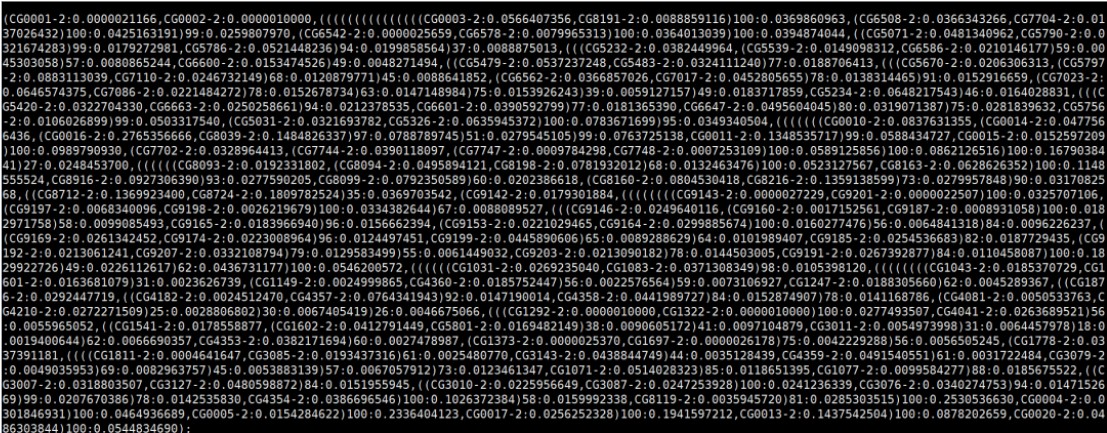
自举树,这里用到了20个,前面为行号(文件本身没有,通过less -SN调出):
分类文件:
2. 绘图
R脚本如下:R包ggtree、ggplot2
# 加载依赖包(未安装则先执行:install.packages(c("ggtree", "ggplot2")))
library(ggtree)
library(dplyr)
# 1. 读取树文件
consensus_tree <- read.tree("iqtree.treefile") # 单棵树
bootstrap_trees <- read.tree("test.nwk") # 多棵树(列表)
# 2. 绘制单棵树
p <- ggtree(consensus_tree,
layout = "slanted", # 树形结构,分支呈斜线
size = 0.3, # 绘图线条粗细
color = "black")
# 3. 叠加自举树(灰色半透明线条)
for (tree in bootstrap_trees) {
p <- p +
geom_tree(
data = tree,
layout = "slanted",
color = "gray90", # 数字大颜色浅
alpha = 0.3, # 透明度设置,值越小越淡
size = 0.5)
}
# 4. 打印出图
print(p)
图片预览如下:
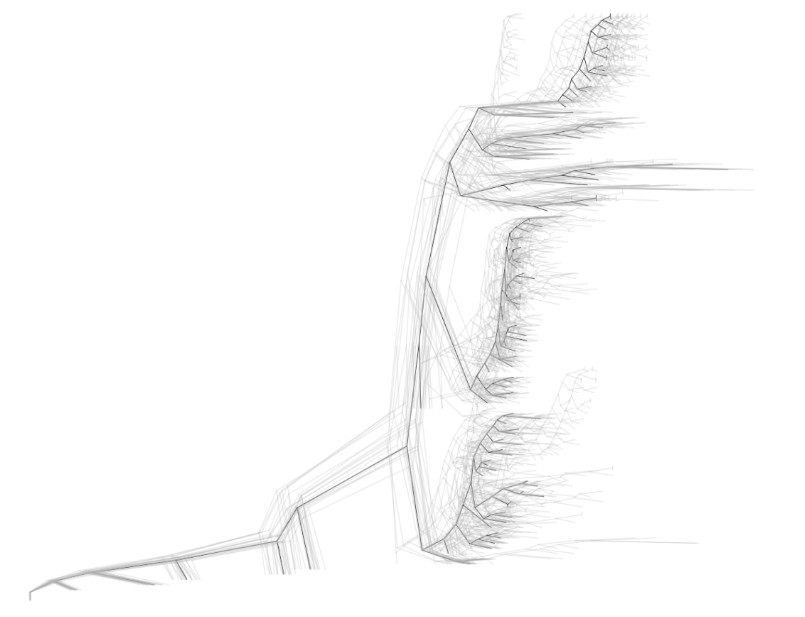
3. 延伸
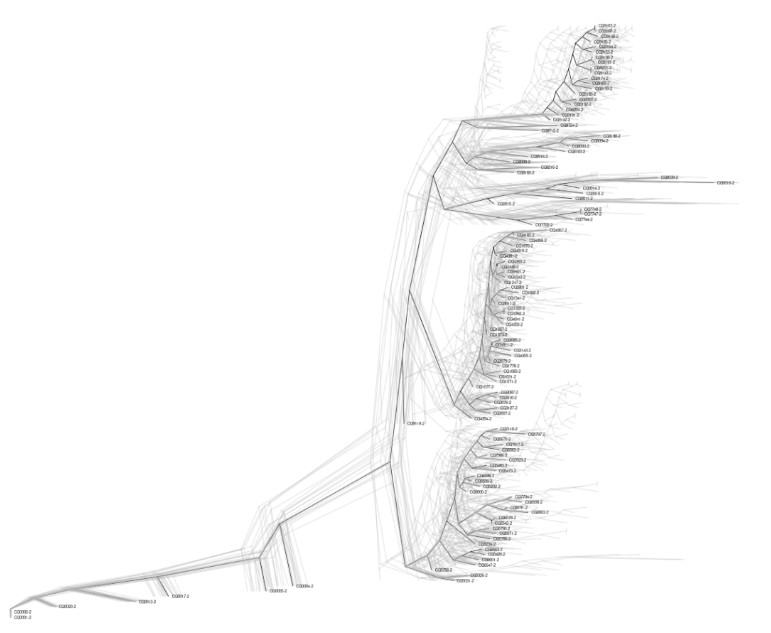
3.1 增加叶子节点
p <- p + geom_tiplab(linesize = 0.02,size=1)
print(p)
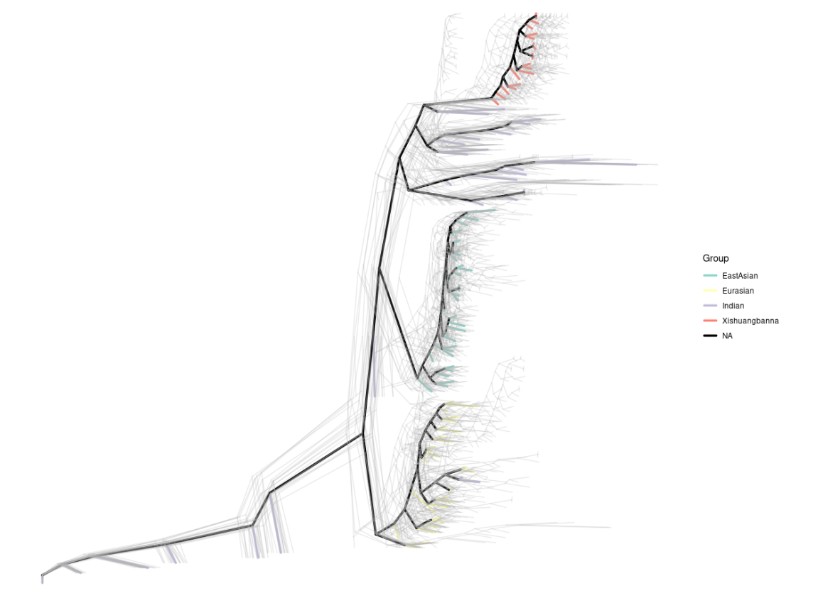
3.2 增加组别颜色关联(dplyr)
library(dplyr) # 用于数据连接
p2 <- ggtree(consensus_tree, layout = "slanted", size = 0.3, color = "black") %<+% group_data +
geom_tree(aes(color = group),size = 1.2,lineend = "round",layout = "slanted") + scale_color_brewer(palette = "Set3",name = "Group",na.value = "black")
for (tree in bootstrap_trees) {
p2 <- p2 +
geom_tree(
data = tree,
layout = "slanted",
color = "gray70",
alpha = 0.3, # 透明度设置,值越小越淡
size = 0.5
)
}
print(p2)
3.3 写成传参的R文件
#!/usr/bin/env Rscript
# 加载必要的包
library(optparse)
library(ggtree)
library(ggplot2)
# 创建参数解析器
option_list <- list(
make_option(c("-w", "--workdir"), type = "character", default = getwd(),
help = "工作目录路径 [默认: 当前目录]",
metavar = "character"),
make_option(c("-c", "--consensus"), type = "character", default = "iqtree.treefile",
help = "共识树文件名 [默认: %default]",
metavar = "character"),
make_option(c("-b", "--bootstrap"), type = "character", default = "iqtree.ufboot",
help = "自举树文件名 [默认: %default]",
metavar = "character"),
make_option(c("-o", "--output"), type = "character", default = "phylogenetic_tree",
help = "输出文件前缀 [默认: %default]",
metavar = "character"),
make_option(c("-l", "--layout"), type = "character", default = "slanted",
help = "树布局类型 (slanted, rectangular, circular, roundrect) [默认: %default]",
metavar = "character")
)
# 解析参数
opt_parser <- OptionParser(option_list = option_list)
opt <- parse_args(opt_parser)
# 设置工作目录
setwd(opt$workdir)
cat("工作目录设置为:", opt$workdir, "\n")
# 检查输入文件是否存在
if (!file.exists(opt$consensus)) {
stop("错误: 共识树文件 '", opt$consensus, "' 不存在!")
}
if (!file.exists(opt$bootstrap)) {
stop("错误: 自举树文件 '", opt$bootstrap, "' 不存在!")
}
cat("正在读取共识树文件:", opt$consensus, "\n")
consensus_tree <- read.tree(opt$consensus)
cat("正在读取自举树文件:", opt$bootstrap, "\n")
bootstrap_trees <- read.tree(opt$bootstrap)
cat("开始绘制进化树...\n")
# 绘制基础树
p <- ggtree(consensus_tree, layout = opt$layout, size = 0.3, color = "black")
# 叠加自举树
for (tree in bootstrap_trees) {
p <- p +
geom_tree(
data = tree,
layout = opt$layout,
color = "gray70",
alpha = 0.3,
size = 0.5
)
}
# 添加叶子标签
p1 <- p + geom_tiplab(linesize = 0.02, size = 1.5)
# 保存图形
output_pdf <- paste0(opt$output, ".pdf")
output_png <- paste0(opt$output, ".png")
cat("保存PDF文件:", output_pdf, "\n")
ggsave(output_pdf, p1, width = 12, height = 10, dpi = 300)
cat("保存PNG文件:", output_png, "\n")
ggsave(output_png, p1, width = 12, height = 10, dpi = 300)
cat("绘图完成! 输出文件:\n")
cat(" -", output_pdf, "\n")
cat(" -", output_png, "\n")
运行如下:
Rscript plot_phylogenetic_tree.R -w ./test -c iqtree.treefile -b test.nwk -o my_tree -l slanted
- 发表于 2025-12-18 14:49
- 阅读 ( 741 )
- 分类:R
