deepseek绘图之--多组学相关性热图
在多组学分析中,经常需要绘制多组学相关性热图,用于展示不同组学数据(如转录组学、蛋白组学、代谢组学、微生物组学等)之间的相关性关系。这种热图可以帮助研究者发现不同层次生物分子之间的关联模式。然而,传统工具如R、Python的绘图流程往往涉及繁琐的文件处理、颜色搭配调试和图例优化等,让研究者耗费大量时间,更是令生信新手望而生畏。AI大模型的介入彻底改变了这一局面。只需提供您的不同组学数据,并用自然语言描述定制需求,AI即可自动生成绘图脚本,真正实现"专业绘图,一键生成"。本文将以蛋白与微生物组为例,带您体验如何通过AI协作,在短短5分钟内完成一幅专业的多组学相关性热图,助您在科研工作中以极致的效率和专业度获得竞争优势!初步构建绘图提示词
要绘制蛋白与微生物相关性热图,为了让他更高效的出图,我们尽量描述的详细一点,这样能够减少我们后面修改的次数,更容易得到想要的结果。首先确认自己的路径和绘图需要的文件,分别是:
蛋白质定量矩阵
 微生物丰度矩阵
微生物丰度矩阵
这里只截取了一小部分,类似的文件大家肯定见过,这里就不多赘述了,
写完了的提示词如下,大家可以参考一下格式:
------------------------------------------
读取两个文件(用read.table读取)
工作路径: /share/nas1/wangq/project/test
蛋白质定量矩阵: pep.txt(行=蛋白质,列=样本)
微生物ASV矩阵:ASV.txt (行=ASV,列= 样本)
计算蛋白质和微生物之间的皮尔森相关性,并使用corrplot绘制相关性热图
绘图工具: corrplot
图形类型:相关性热图
------------------------------------------
在deepseek输出上面写好的提示词,如果问题复杂可以打开deepthink,
这样简单的就不需要了。
初步生成绘图代码

AI给出了详细的代码,我们来到RStudio运行一下,结果如下:
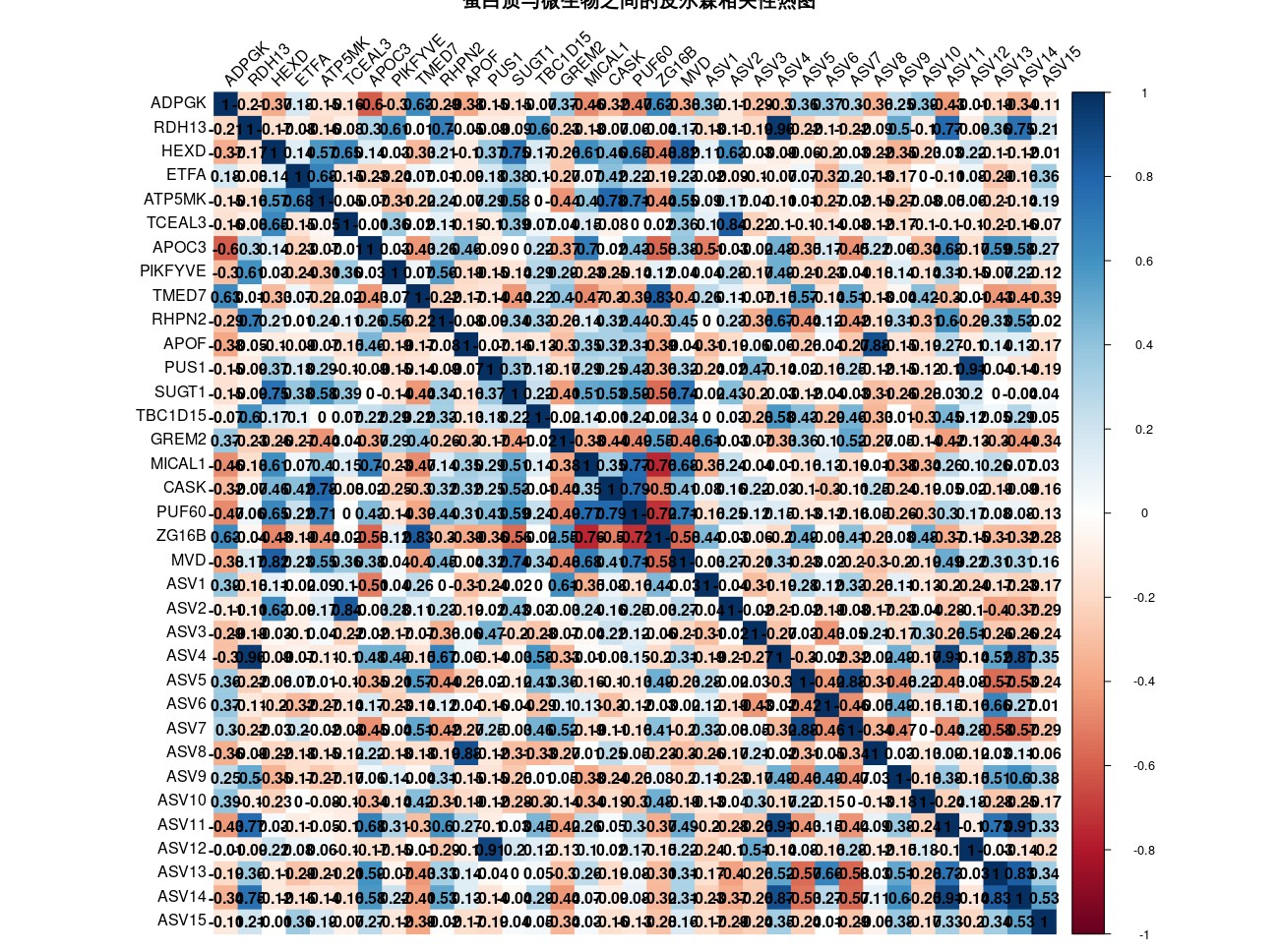
可以看到它把蛋白与微生物合并一起计算相关性了,我们让他修改一下不合并。
让AI帮助我修改可视化效果

修改后的绘图结果如下所示:
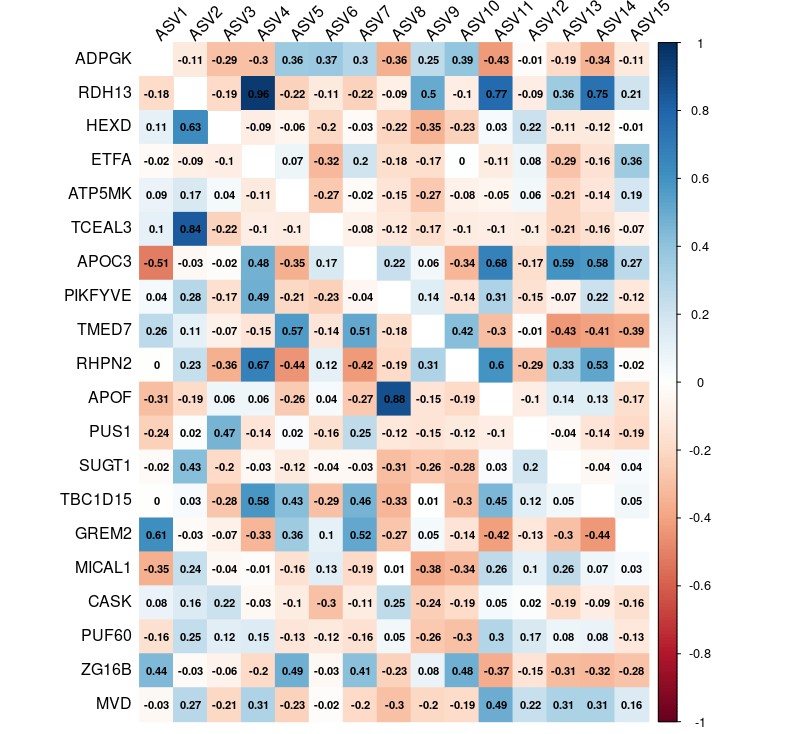
这里使用颜色表示相关性的强度,我们可以让他改成使用圆圈大小,并去掉相关性值的显示。

修改后的绘图如下所示,比较符合我们的要求,如果有其他需求,也可以让他再调。
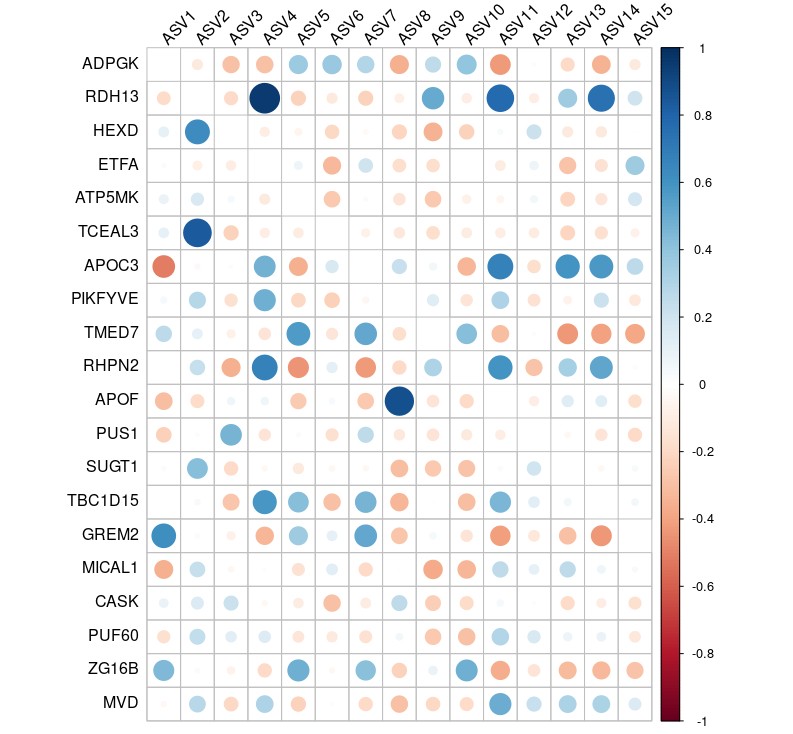
最终绘图代码
# 设置工作路径setwd("/share/nas1/wangq/project/test")# 加载所需库library(corrplot)# 读取数据(保持原始结构:行=特征,列=样本)protein <- read.table("pep.txt", header = TRUE, row.names = 1, sep = "\t", check.names = FALSE)microbe <- read.table("ASV.txt", header = TRUE, row.names = 1, sep = "\t", check.names = FALSE)# 筛选共同样本并保持顺序一致common_samples <- intersect(colnames(protein), colnames(microbe))protein <- protein[, common_samples, drop = FALSE]microbe <- microbe[, common_samples, drop = FALSE]# 转置为样本×特征格式(便于计算相关性)protein_samples <- t(protein) # 行=样本,列=蛋白质microbe_samples <- t(microbe) # 行=样本,列=微生物# 直接计算两组特征间的交叉相关性(不合并数据)# 结果矩阵:行=蛋白质,列=微生物correlation_results <- cor(protein_samples, microbe_samples, method = "pearson")# 绘制热图corrplot( correlation_results, method = "circle", type = "full", tl.col = "black", tl.srt = 45, diag = FALSE # 交叉矩阵无对角线意义)
更多生信课程:
- 发表于 2025-07-30 18:18
- 阅读 ( 4100 )
- 分类:R
