在微生物组研究中,揭示群落组成与动态变化是核心任务。无论是16S rRNA测序、宏基因组分析,还是环境样本的物种丰度比较,研究者常需面对一个关键问题:如何直观展示多个样本在不同分类水平(如...
在微生物组研究中,揭示群落组成与动态变化是核心任务。无论是16S rRNA测序、宏基因组分析,还是环境样本的物种丰度比较,研究者常需面对一个关键问题:如何直观展示多个样本在不同分类水平(如门、属、种)上的物种相对丰度?堆叠柱状图以其“多维度信息整合”的优势,成为生信论文中的经典可视化工具,通过颜色区分物种、柱高表示丰度,清晰呈现样本间的群落结构差异。然而,传统工具如R(ggplot2, phyloseq)或Python(matplotlib, seaborn)的绘图流程往往涉及复杂的数据预处理、颜色搭配调试和图例优化,耗费时间,令生信新手望而生畏。
AI大模型的出现彻底颠覆了这一困境。只需上传你的OTU丰度表或物种相对丰度矩阵(如CSV格式,包含样本和分类单元),并用自然语言描述分组信息(如不同采样点、处理条件或时间序列),AI即可自动生成“端到端”绘图脚本:从数据归一化、物种筛选,到自定义颜色梯度(适配高丰度/低丰度物种)、柱状堆叠顺序优化,再到生成可交互的高分辨率堆叠柱状图——图例自动排布,样本分组清晰标注,配色符合期刊发表标准,一切“所想即所得”。
本文将带你体验如何通过AI协作,在短短5分钟内完成一幅精美的微生物堆叠柱状图,助你在微生物组研究赛道上以极致的分析速度和视觉表现力脱颖而出!
初步构建绘图提示词
需要绘制微生物堆叠柱状图,为了让他更高效的出图,我们尽量描述的详细一点,这样能够减少我们后面修改的次数更容易得到想要的结果,首先确认自己的路径和绘图需要的文件,需要3个文件,做相关分析的同学肯定不陌生分别是
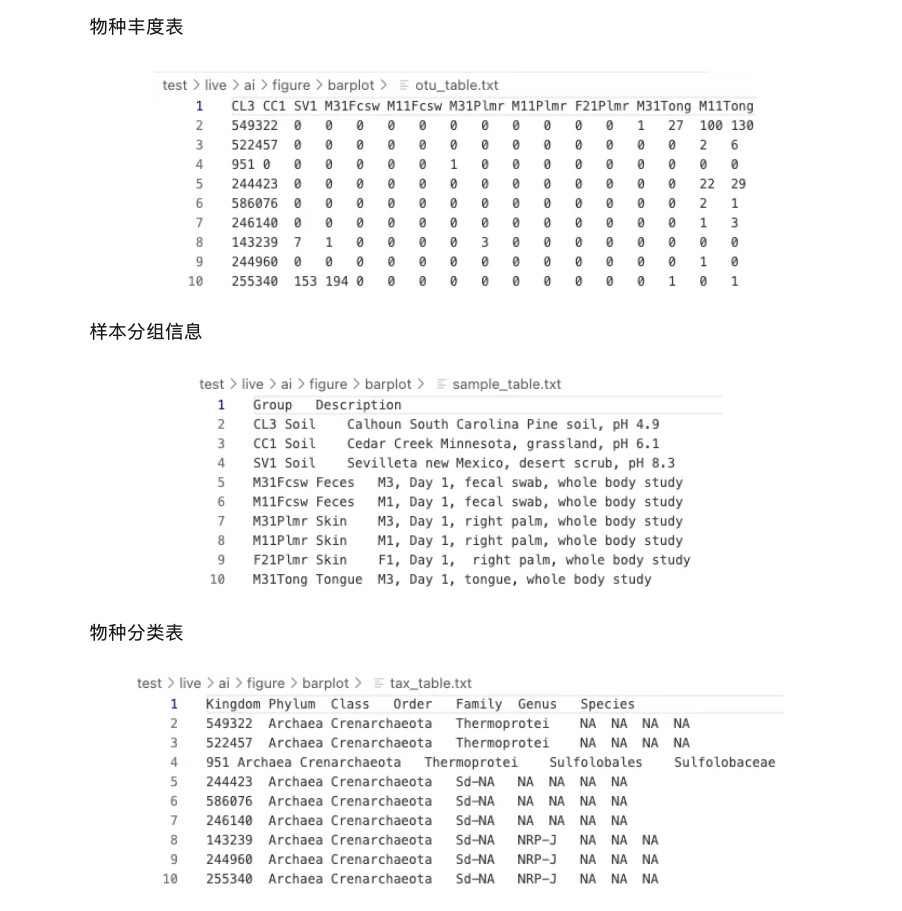
一般做微生物相关分析都需要这三个文件,有的地方分类信息和丰度表会组合成一个表,如果是那样就需要改一下提示词,尽可能清楚的描述
写完了的提示词如下,大家可以参考一下格式
------------------------------------------
读取3个文件(TSV格式,用read.delim读取)
工作路径: /home/wanghj/test/live/ai/figure/barplot
物种丰度矩阵:otu_table.txt(行=物种,列=样本)
分类学信息表:tax_table.txt(含7个层级分类单元)
样本元数据:sample_table.txt(含分组信息列)
使用tidyverse处理数据,过滤掉丰度低于1/10000的物种,只绘制丰度最高的前15个,其余的归到other,用门级别进行绘图
绘图工具:ggplot2
图形类型:相对分度的堆叠柱状图,仅绘制不输出
颜色映射:RColorBrewer的set3
坐标轴:坐标轴展示左下,柱子贴住x轴
主题:隐藏网格线
------------------------------------------
在deepseek输出上面写好的提示词,如果问题复杂可以打开deepthink
这样简单的就不需要了
初步生成绘图代

给了详细的代码,我们来到rstudio运行一下结果如下

这明显出了问题,柱子不一样高,检查中文注释得知,他先计算了相对丰度在过滤,后面没有重新算,导致物种的丰度总和不为1了,问题描述清楚让他重新生成
让AI我修改可视化效果
 得到的结果如下,完全符合要求,如果有其他需求比如颜色啥的,也可以让他调
得到的结果如下,完全符合要求,如果有其他需求比如颜色啥的,也可以让他调

# 加载必要的包
library(tidyverse)
library(RColorBrewer)
# 设置工作路径
setwd("/home/wanghj/test/live/ai/figure/barplot")
# 读取数据
otu_table <- read.delim("otu_table.txt", row.names = 1, check.names = FALSE)
tax_table <- read.delim("tax_table.txt", row.names = 1)
sample_table <- read.delim("sample_table.txt", row.names = 1)
# 数据处理流程(先过滤低丰度物种,再计算相对丰度)
plot_data <- otu_table %>%
# 转换为长格式
rownames_to_column(var = "OTU") %>%
pivot_longer(cols = -OTU, names_to = "Sample", values_to = "Abundance") %>%
# 添加分类信息(Phylum级别)
left_join(tax_table %>% rownames_to_column(var = "OTU"), by = "OTU") %>%
# 按样本计算相对丰度(先不过滤)
group_by(Sample) %>%
mutate(RelativeAbundance = Abundance / sum(Abundance)) %>%
ungroup() %>%
# 过滤低丰度物种(<1/10000)
filter(RelativeAbundance > 0.0001) %>%
# 重新计算相对丰度(确保总和为1)
group_by(Sample) %>%
mutate(RelativeAbundance = Abundance / sum(Abundance)) %>%
ungroup() %>%
# 按Phylum汇总
group_by(Sample, Phylum) %>%
summarise(RelativeAbundance = sum(RelativeAbundance), .groups = "drop") %>%
# 选择丰度最高的15个Phylum,其余归为"Other"
group_by(Phylum) %>%
mutate(TotalAbundance = sum(RelativeAbundance)) %>%
ungroup() %>%
mutate(Phylum = ifelse(Phylum %in% head(unique(Phylum[order(-TotalAbundance)]), 15),
Phylum, "Other")) %>%
# 重新汇总(确保Other合并正确)
group_by(Sample, Phylum) %>%
summarise(RelativeAbundance = sum(RelativeAbundance), .groups = "drop") %>%
# 添加分组信息
left_join(sample_table %>% rownames_to_column(var = "Sample"), by = "Sample")
# 绘图(确保柱子高度一致)
ggplot(plot_data, aes(x = Sample, y = RelativeAbundance, fill = Phylum)) +
geom_col(position = "stack", width = 0.7) +
scale_fill_manual(values = colorRampPalette(brewer.pal(12, "Set3"))(length(unique(plot_data$Phylum)))) +
labs(x = NULL, y = "Relative Abundance") +
theme_minimal() +
theme(
panel.grid = element_blank(),
axis.line = element_line(color = "black"),
axis.ticks = element_line(color = "black"),
axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "right"
) +
scale_y_continuous(expand = c(0, 0)) # 确保柱子贴住x轴
更多生信课程:
