mclust analysis.r 基于模型的聚类
R包mclust对样本进行聚类
使用方法:
$Rscript ../scripts/mclust_analysis.r -h
usage: ../scripts/mclust_analysis.r [-h] -i gene_data -m metadata [--mclust]
[-g group] [-n model_name] [-o outdir]
[-p prefix]
mclust analysis:https://www.omicsclass.com/article/1580
optional arguments:
-h, --help show this help message and exit
-i gene_data, --gene_data gene_data
input data file path[required]
-m metadata, --metadata metadata
input clinical information file path[required]
--mclust whether to cluster the samples[optional,default:False]
-g group, --group group
Group the samples into several
categories[optional,default:3]
-n model_name, --model_name model_name
input the model category to use[optional,default:VVE]
-o outdir, --outdir outdir
output file directory[optional,default cwd]
-p prefix, --prefix prefix
out file name prefix[optional,default metadata]
参数说明:
-i 输入基因的表达数据:
| ID | TCGA-A3-3319-01A-02R-1325-07 | TCGA-A3-3323-01A-02R-1325-07 |
| YTHDC2 | 16.5128725081007 | 20.6535652352011 |
| ELAVL1 | 44.3876796198438 | 31.8729000784291 |
-m 输入样本的临床信息:
| barcode | patient | TCGA_Study |
| TCGA-A3-3319-01A-02R-1325-07 | TCGA-A3-3319 | KIRC |
| TCGA-A3-3323-01A-02R-1325-07 | TCGA-A3-3323 | KIRC |
--mclust 是否对样本进行聚类
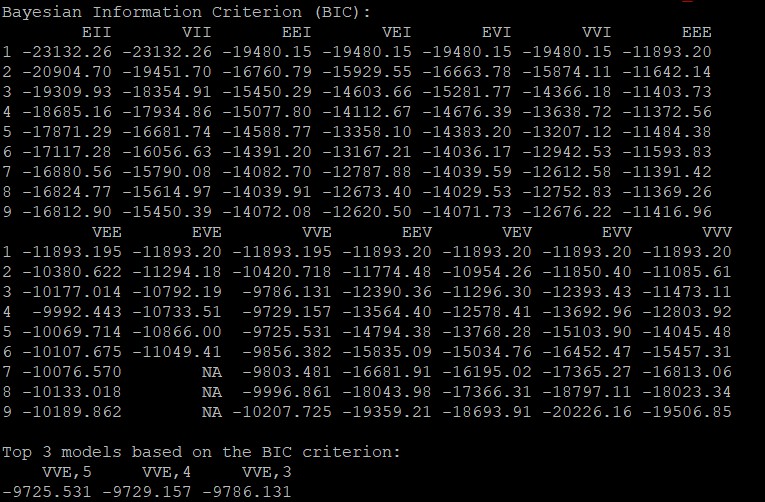
第一次运行脚本不进行聚类,通过返回的BIC(贝叶斯信息判别标准)结果选择合适的聚类数和模型类别
-n 输入选择的模型类别
mclust包中提供了14种模型(EII、VII、EEI、VEI、EVI、VVI、EEE、EVE、VEE、VVE、EEV、VEV、EVV、VVV)
-g 输入合适的聚类数
使用举例:
#第一次运行不指定g和n,通过BIC结果选择合适的g和n
$Rscript ../scripts/mclust_analysis.r -i m6a_gene_TPM.tsv -m ../metadata_surv_immu.tsv
#再次运行指定g和n,进行聚类
Rscript ../scripts/mclust_analysis.r -i m6a_gene_TPM.tsv \
-m ../metadata_surv_immu.tsv --mclust -g 3 -n VVE
结果展示:
含有样本聚类结果的文件:metadata_group.tsv
- 发表于 2021-10-22 15:33
- 阅读 ( 3268 )
- 分类:R
