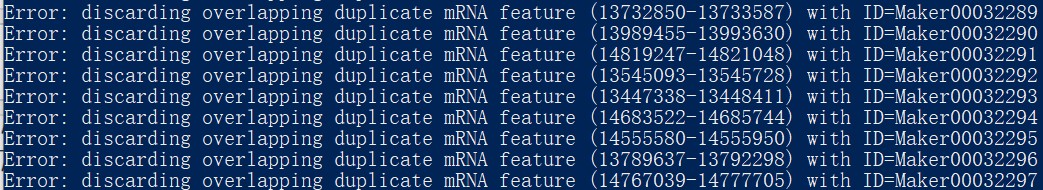
gff文件中缺少基因gene注释信息行,导致gtf文件不标准,出现报错KeyError: 'gene_id',使得后续无法获取gene_length.txt文件。可以重新找一个存在gene注释信息的基因组。也可以人为添加上相关信息,参考笔记:https://www.omicsclass.com/article/2081
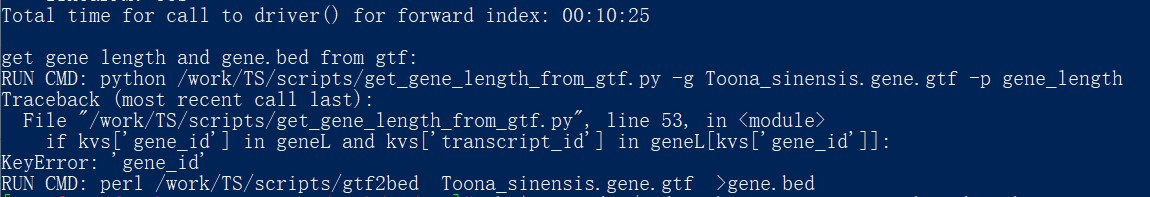
RNAseq有参转录组数据自主分析课程 ,运行建立基因组索引命令时,报错,无法获取gene_length.txt文件,请问是什么原因?如何处理呢?
根据RNAseq有参转录组数据自主分析课程 ,运行建立基因组索引命令时,报错,无法获取gene_length.txt文件,请问是什么原因?如何处理呢?另外,得到的exons.tsv和splicesites.tsv文件是空的,请问这种情况正常吗?
运行代码:sh $scriptdir/index.sh Toona_sinensis.genome.fasta Toona_sinensis.gene.gff
报错信息如下:get gene length and gene.bed from gtf:
RUN CMD: python /work/TS/scripts/get_gene_length_from_gtf.py -g Toona_sinensis.gene.gtf -p gene_length
Traceback (most recent call last):
File "/work/TS/scripts/get_gene_length_from_gtf.py", line 53, in <module>
if kvs['gene_id'] in geneL and kvs['transcript_id'] in geneL[kvs['gene_id']]:
KeyError: 'gene_id'
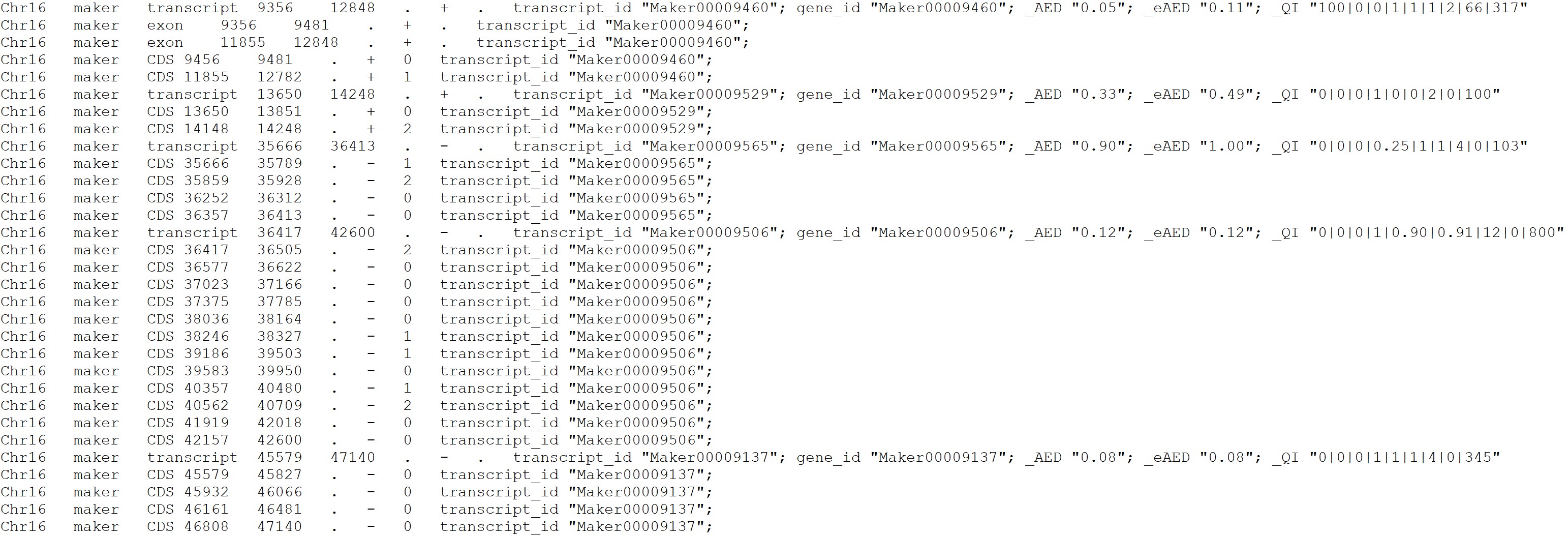
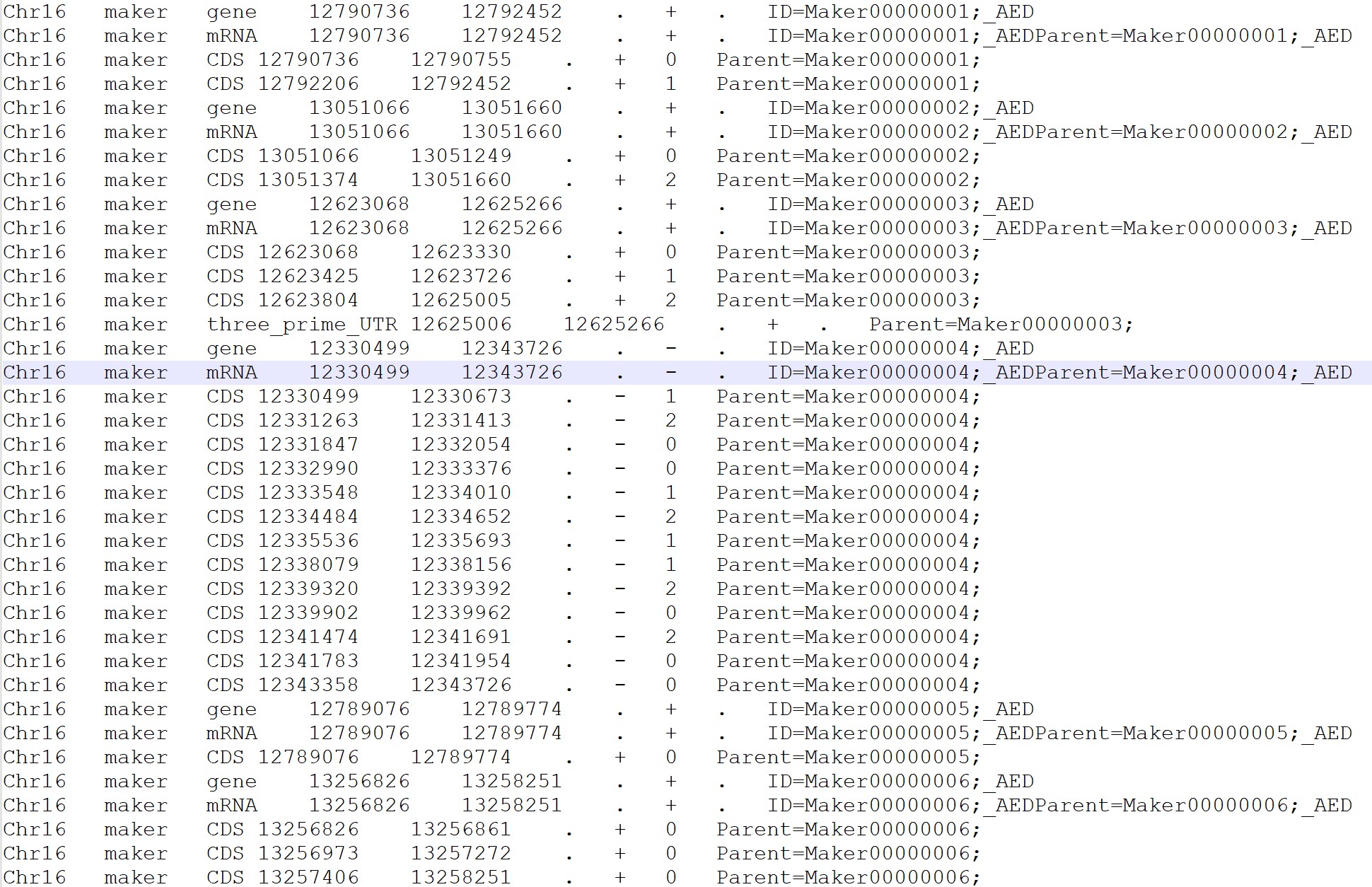
基因组gff文件如下:
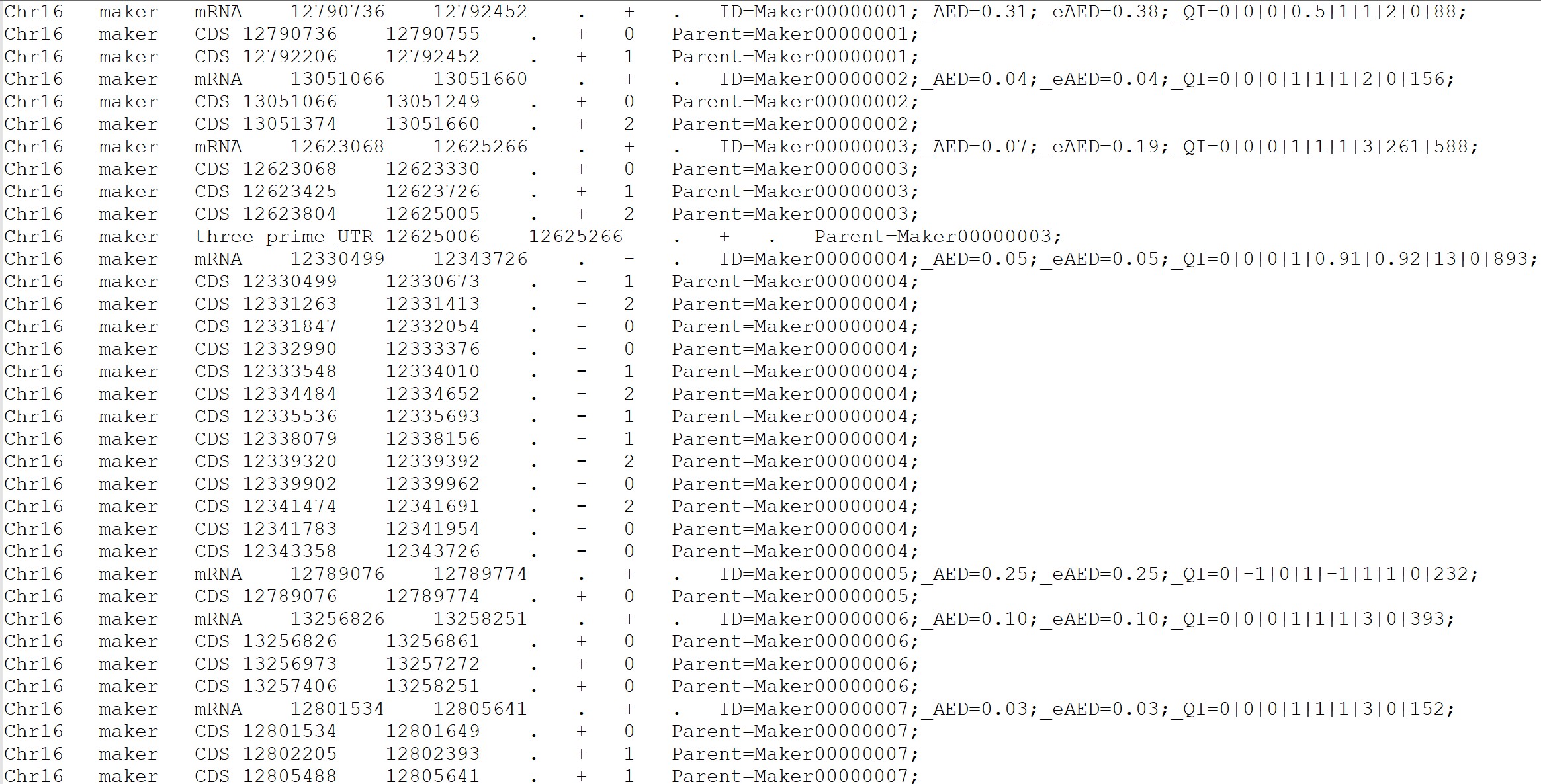
gff转化得到的gtf文件如下:
2 个回答
添加gene注释行后的gff文件如下:
 报错信息如下:get gene length and gene.bed from gtf:
报错信息如下:get gene length and gene.bed from gtf:
RUN CMD: python /work/TS/scripts/get_gene_length_from_gtf.py -g 0Toona_sinensis.gene.gtf -p gene_length
Traceback (most recent call last):
File "/work/TS/scripts/get_gene_length_from_gtf.py", line 53, in <module>
if kvs['gene_id'] in geneL and kvs['transcript_id'] in geneL[kvs['gene_id']]:
KeyError: 'gene_id'
RUN CMD: perl /work/TS/scripts/gtf2bed 0Toona_sinensis.gene.gtf >gene.bed